
Vol. 37, n.º 2, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 2, 2004 |
Olga Balagué, Lluis Colomo, Elías Campo
Unidad de Hematopatología, Servicio de Anatomía Patológica, Hospital Clinic, Institut d’Investigacions Biomèdiques August Pi I Sunyer (IDIBAPS), Universitat de Barcelona ECAMPO@clinic.ub.es
RESUMEN
El linfoma de células del manto (LCM) es un subtipo de linfoma no Hodgkin de fenotipo B que representa entre un 2’5 y un 10% de todos los linfomas. Histológicamente puede presentar un patrón de crecimiento en la zona del manto, nodular o difuso y se diferencian dos variedades, típica y blastoide. Clínicamente, suele presentarse en varones de edad avanzada, de forma diseminada y con frecuente afección extranodal. Fenotípicamente se caracteriza por la presencia de una población monoclonal de línea B con positividad para CD5 y sobreexpresión de ciclina D1asociada a la presencia constante de la translocación t(11;14). En las formas típicas y blastoides se han identificado frecuentemente alteraciones en el gen ATM. Las formas blastoides presentan además alteraciones genéticas adicionales que incluyen inactivación de los genes supresores tumorales p53 y p16INK4a. La supervivencia media de LCM oscila entre 3 y 5 años.
Palabras clave: linfoma de células del manto, ciclina D1
SUMMARY
Mantle-cell lymphoma (MCL) is a B-cell non-Hodgkin lymphoma representing 2.5 to 10% of lymphomas. It is characterized by a mantle zone, nodular, or diffuse pattern of growth, Two cytological variants have been identified: typical and blastoid. Clinically, MCL presents in elderly males with advanced disease and frequent extranodal involvement. Phenotypically, MCL is characterized by a monoclonal B-cell population, positive for CD5 and cyclin D1 overexpression. Cyclin D1 is not normally expressed in lymphocytes or myeloid cells but it is constantly expressed in MCL indicating an important role in the pathogenesis of this lymphoma. The characteristic cytogenetic alteration in MCL is the t(11;14), leading to the overexpression of cyclin D1, implicated in the deregulation of the cell cycle. Additional alterations of ATM have been identified in the typical and blastoid variants. In the blastoid variants, inactivation of p53 and p16INK4a tumor-suppressor genes has been observed. The median survival of MCL is 3-5 years.
Key words: mantle-cell lymphoma, cyclin D1.
INTRODUCCIÓN
El linfoma de células del manto (LCM) es un proceso linfoproliferativo derivado de un subgrupo de células B, en un estadio previo al paso por el centro germinal. Se caracteriza por una proliferación monomorfa de linfocitos de tamaño entre pequeño e intermedio, con núcleos irregulares que coexpresan CD5. Genéticamente muestra reordenamiento de bcl-1, con una sobreexpresión constante de ciclina D1. El comportamiento biológico del LCM es muy agresivo, con una supervivencia media de 3-5 años, pero la gran mayoría de pacientes no pueden considerarse curados.
Epidemiología y manifestaciones clínicas
El LCM representa del 2.5 al 10% de todos los linfomas no Hodgkin, ocurre en individuos de edad avanzada, con una media de 60 años y predominantemente en varones. El 70% de los pacientes se presentan en estadio avanzado, con linfadenopatía generalizada y afectación de médula ósea. La hepatomegalia y la esplenomegalia son relativamente frecuentes, esta última asociada en muchos casos a la afectación de sangre periférica. También es frecuente la afectación extranodal, pero sólo en un 4-15% de casos en ausencia de afectación nodal. La afectación gastrointestinal se ha reportado en el 10-25% de los pacientes, tanto en el momento de la presentación como durante el curso de la enfermedad, en cambio la afectación del sistema nervioso central, que puede ocurrir en el 10-20% de los casos suele ser un evento tardío en la evolución de la neoplasia. La expresión en sangre periférica varía del 20 al 58% de los casos. Algunos pacientes pueden presentarse con una forma leucémica muy agresiva, imitando una leucemia aguda. Estos casos suelen mostrar una morfología blastoide, con cariotipos complejos y una evolución muy rápida. La detección de una segunda neoplasia, de forma previa, simultánea o posterior al diagnóstico de LCM, se ha descrito en el 12-21% de los pacientes.
Célula de origen y bases patogénicas moleculares
LCM es un proceso linfoproliferativo derivado de una población de células B que no han pasado por el centro germinal y que expresan CD5. Las células B humanas CD5 positivas están presentes en los tejidos linfoides fetales y en la sangre, disminuyendo con la edad. En los adultos, las células B CD5 positivas, circulan en pequeño número y se localizan en los folículos primarios y en la zona del manto de los folículos secundarios. La expresión de CD5 en el LCM es muy alta, recordando la intensidad que se observa en las células B fetales, en contraste con los bajos niveles detectados en las células del manto folicular de los adultos. El LCM también mantiene la expresión de diversos genes que se expresan normalmente en las células vírgenes y en las células de la zona del manto. Los estudios inmunológicos y la identificación de muy pocas o ninguna mutación somática en los genes de las inmunoglobulinas en la mayoría de LCM han confirmado su origen en células previas al centro germinal. Además, análisis recientes de los puntos de ruptura de JH/bcl-1, han sugerido que esta translocación se genera predominantemente en un reordenamiento inicial de DH-JH en células B tempranas. Estudios recientes demuestran que un 20-30% de LCM muestra hipermutaciones somáticas de los genes de las inmunoglobulinas, con una tendencia a la utilización del gen VH4-34, un gen utilizado preferentemente por las células B involucradas en fenómenos autoinmunes, sugiriendo que estos tumores podrían originarse en poblaciones específicas de células autoreactivas B.
Translocación t(11;14) y sobreexpresión de ciclina D1
La translocación t(11;14)(q13;q32) está presente en prácticamente todos los casos de LCM. El gen de ciclina D1 se yuxtapone a la región de unión (J) de la cadena pesada de las inmunoglobulinas (IgH), determinando una expresión anómala de ciclina D1 en las células linfoides. El 35% de las rupturas de 11q13 ocurren en una zona denominada cluster mayor de translocación (MTC), que se puede amplificar por técnicas de PCR. Por tanto mediante esta técnica sólo se detectan el 35% de las translocaciones, mientras que por técnica de hibridación in situ con fluorescencia se detectan prácticamente todos los casos. La translocación t(11;14) se ha identificado en ocasionales LLC atípicas, en el 20-30% de las leucemias prolinfocíticas y en el 5% de mielomas múltiples. El análisis molecular de la translocación en el LCM y en el mieloma múltiple sugiere que el mecanismo debe ser diferente en los dos tumores. Además la amplificación del gen de la ciclina D1 sin translocación se ha documentado en el mieloma múltiple pero no en el LCM.
Las ciclinas tipo D junto con las kinasas 4 y 6 dependientes de ciclina (cdk) regulan la transición celular entre la fase G1 y S del ciclo, mediante la fosforilación de la proteína del retinoblastoma (Rb). Durante la fase G1 del ciclo, estímulos extracelulares determinan el paso de la célula a la siguiente fase. En el LCM, ciclina D1 se expresa de forma continua e independiente de los factores externos, además ciclina D1 se une al Rb y puede vencer su efecto supresor independientemente del estado de fosforilación. En las células normales dos tipos de inhibidores regulan los complejos ciclina-cdk: el tipo cip/kip que incluye p21WAF1 y p27KIP1, e inhibe las ciclinas D y E, y la familia INK4 que incluye p16INK4A e inhibe selectivamente las kinasas asociadas a ciclina D. El gen que codifica p16INK4A , también codifica para una segunda proteína, p14ARF que estabiliza la proteína p53 , bloqueando su degradación mediada por MDM2. Por tanto, el locus INK4a/ARF es un enlace regulador común entre las vías de p53 y p16INK4A y a través de esta última de Rb. La inactivación de p53 por mutación o delección es más frecuente en las variantes blásticas, sin embargo como p53 es activada por ATM en respuesta al daño del DNA, la disminución de la función de p53 puede deberse también a la pérdida de ATM que se da en el LCM. La detección de p27 está disminuida en el LCM como resultado de un aumento de la degradación proteica mediada por proteosoma y por su unión a ciclina D1.
ATM y delección de 11q
Además de la translocación t(11;14), una de las alteraciones cromosómicas más frecuentes es la delección de la región11q22-23, incluyendo un área donde se encuentra el gen ATM. El gen ATM codifica para una kinasa serin-treonina, que juega un papel importante en las vías de respuesta al daño del DNA representado por rupturas de la doble cadena y evita que la célula continúe su división en presencia de daño importante del DNA. Mutaciones de ATM se han detectado en 46-67% de leucemias prolinfocíticas T (LPL-T), 19-34% de las LLC-B y más recientemente en 40-75% de los LCM. La inactivación de ATM ocurre tanto en el LCM típico como en el blastoide. También se han detectado mutaciones de ATM en la línea germinal y en células previas a la diferenciación linfoide en pacientes con LLC y LCM, señalando la posibilidad de que las mutaciones de ATM sean un evento predisponente muy temprano. Una de las dianas de la proteína ATM es p53. En el LCM las mutaciones de p53 ocurren en tumores con ATM mutada y no mutada, lo que sugiere que estas dos alteraciones pueden actuar de forma independiente en la patogénesis del tumor.
Morfología
Ganglio linfático
Patrones arquitecturales
Los ganglios linfáticos afectados muestran una arquitectura borrada por una proliferación linfoide atípica, que puede adoptar tres patrones de crecimiento: zona del manto, nodular o difuso (fig. 1). El patrón de zona del manto se caracteriza por una expansión del área del manto por células tumorales que rodean centros germinales «desnudos» reactivos. Este patrón puede estar asociado con una preservación parcial de la arquitectura nodal y puede ser difícil de distinguir de una hiperplasia folicular o de células del manto. Son comunes las áreas de transición entre los patrones nodular y difuso pero, en raros casos, la nodularidad puede ser prominente y llevar a un diagnóstico erróneo de linfoma folicular. Algunos nódulos pueden ser sólidos, sin evidencia de centros germinales residuales y podrían representar la contrapartida maligna de los folículos primarios. De forma alternativa, el patrón nodular puede ser debido a una infiltración masiva y una obliteración de los centros germinales originales por las células tumorales. En algunos casos la tinción con ciclina D1 puede ayudar a reconocer la infiltración inicial o la colonización de los centros germinales reactivos que puede corresponder a los estadios tempranos de un patrón nodular. Centros germinales residuales se pueden ver también en tumores con un patrón más difuso, aunque en esos casos sólo se identifican focalmente.
![]()
Fig. 1: Patrones arquitecturales en el
linfoma de células del manto. (A) Patrón de zona del manto: Las células
tumorales expanden la corona de células del manto, que rodea a los centros
germinales reactivos. (B) Patrón nodular: Los nódulos están compuestos por
células tumorales sin evidencia de centros germinales residuales. (C) Patrón
difuso: El ganglio linfático está difusamente infiltrado por las células
tumorales.
Variantes citológicas
Se han descrito dos variantes citológicas principales de LCM: típica y blastoide (tabla 1).

El LCM típico (común o clásico) se caracteriza por una proliferación monótona de linfocitos de tamaño entre pequeño y mediano, con escaso citoplasma, núcleo variablemente irregular, cromatina condensada y nucleolo inconspicuo (fig. 2). La presencia de células grandes con citoplasma abundante o nucleolos prominentes es rara. Cuando están presentes, corresponden a centroblastos reactivos de los centros germinales residuales borrados por las células del linfoma. Casos ocasionales pueden mostrar un predominio de linfocitos pequeños con núcleos redondeados. Contrariamente a los casos típicos, el número de células con núcleos irregulares o indentados es menos prominente. Esta variante puede ser difícil de distinguir de la leucemia linfática crónica/linfoma linfocítico de célula pequeña (LLC/SLL).
![]()
Fig. 2: Variante típica del LCM que se
caracteriza por linfocitos de tamaño pequeño a intermedio, con núcleo irregular,
cromatina condensada y escaso citoplasma. Presencia de algunos histiocitos de
citoplasma eosinófilo.
No se han observado diferencias en el comportamiento clínico entre esta variante de célula pequeña y los casos típicos, pero es importante distinguirlas para evitar un diagnóstico erróneo de LLC (tabla 3). La actividad proliferativa en el LCM típico y de célula pequeña puede variar de un caso a otro, pero habitualmente es menor de 1-2 mitosis por campo de gran aumento. Algunos tumores con una morfología típica pueden mostrar un índice mitótico relativamente alto, parecido a los de la variante blastoide y los pacientes pueden presentar un curso clínico agresivo. Los histiocitos epitelioides con citoplasma eosinófilo son relativamente comunes, pero no se observan microgranulomas. Estos histiocitos generalmente no contienen cuerpos apoptóticos. Se identifica frecuentemente una trama prominente de células dendríticas foliculares con el típico aspecto de núcleos superpuestos, membranas nucleares delicadas y una apariencia de núcleos vacíos. Una trama densa y concéntrica puede representar la colonización de un centro germinal preexistente, mientras que una trama laxa y de patrón irregular puede corresponder a la expansión de folículos primarios.

La variante blastoide es más agresiva y su morfología varía desde una población monótona de células que parecen linfoblastos, a una apariencia más pleomórfica con células irregulares de mayor tamaño. Aunque estos casos son citológicamente distintos del LCM típico, las similitudes del fenotipo y de las alteraciones genéticas, con translocaciones de 11q13, reordenamientos de bcl-1 y sobreexpresión de ciclina D1 han permitido reconocerlos como variantes agresivas de LCM. El término blastoide incluye las variantes blástica y pleomórfica, previamente reconocidas, que pueden representar los extremos de un espectro morfológico, puesto que en algunos casos se pueden observar áreas de transición entre estos subtipos. El LCM blástico se caracteriza por una población monótona de linfocitos de tamaño intermedio con escaso citoplasma, núcleos redondeados con cromatina fina dispersa y nucleolo inconspicuo. Estos casos pueden parecer un linfoma linfoblástico o una afectación nodal por una leucemia mieloide aguda. El índice mitótico es muy elevado, con más de 2-3 mitosis por campo de gran aumento. Se pueden ver histiocitos de cuerpo tingible y un patrón en «cielo estrellado» (fig. 3A).
Los LCM con una morfología pleomórfica o de células grandes fueron designados inicialmente en la clasificación de Kiel como linfomas centrocíticos «anaplásicos» o linfomas centroblásticos de subtipo « centrocítico». Estos tumores están compuestos por una población más heterogénea de células grandes con núcleos indentados de forma ovalada o irregular, cromatina fina dispersa y nucleolo evidente de pequeño tamaño. De forma similar a otros casos blásticos, el índice mitótico es muy elevado. En algunos de estos casos las figuras de mitosis pueden mostrar una marcada hipercromasia. Este hallazgo está habitualmente asociado con la presencia de clones tetraploides en el LCM pleomórfico. Esta variante pleomórfica puede ser difícil de distinguir del linfoma de célula grande (tabla 3). Algunos LCM leucémicos descritos como «variantes prolinfocíticas» del LCM podrían, en realidad, representar formas leucémicas del subtipo pleomórfico del LCM (fig. 3B).
![]()
Fig. 3: Variante blastoide. (A) Las
células tumorales son linfocitos de tamaño intermedio con núcleo redondo y
cromatina finamente dispersa, nucleolo pequeño o inconspicuo y elevado índice
mitótico. (B) Las formas pleomórficas presentan mayor variabilidad en forma y
tamaño nuclear.
Algunos casos pueden tener un número variable de células con citoplasma pálido y más abundante, que recuerdan a las células B monocitoides. El núcleo de estas células puede tener una morfología clásica o blastoide pero el aspecto peculiar del citoplasma puede plantear el diagnóstico diferencial con el linfoma de la zona marginal o la tricoleucemia (tabla 3). En algunos casos estas células de aspecto monocitoide pueden incluso expandirse hacia la zona marginal de los folículos linfoides por fuera de una zona del manto aparentemente preservada (fig. 4).

Fig. 4: LCM con diferenciación marginal.
(A) Patrón de crecimiento de las zonas del manto y marginal a expensas de una
población monótona con citoplasma amplio y claro (B).
Localizaciones extranodales
Médula ósea y sangre periférica
La infiltración de la médula ósea, independientemente de la afectación de sangre periférica ocurre en el 50 al 91% de los pacientes y se detecta más frecuentemente en biopsias de cilindros óseos que en los aspirados. El patrón de infiltración puede ser nodular (fig. 5A), intersticial, o paratrabecular, siendo en la mayoría de los casos una combinación de estos patrones. Los agregados paratrabeculares aislados son extremadamente raros. En algunos casos se puede ver una infiltración difusa de la médula (fig. 5B). El grado de infiltración no parece correlacionar con la variante histológica de LCM identificada en la biopsia de ganglio linfático, el patrón arquitectural o la supervivencia del paciente. Las tinciones de inmunohistoquímica incluyendo ciclina D1 y p27 pueden utilizarse en el diagnóstico diferencial de biopsias de médula ósea, para distinguir la afectación por LCM de otros linfomas de célula pequeña.

Fig. 5: (A) Patrón nodular de infiltración
de la médula ósea. (B) Patrón difuso. (C) Citología de LCM típico en sangre
periférica.
La apariencia citológica de las células tumorales en sangre periférica y en aspirados de médula ósea muestra un espectro similar al de las muestras de tejido. Las células circulantes en la mayoría de LCM, habitualmente son una mezcla de linfocitos de tamaño pequeño e intermedio con escaso citoplasma, irregularidades nucleares prominentes y cromatina reticular (fig. 5C). Algunas células pueden tener núcleos redondeados, pero la cromatina no tiene el aspecto grumoso que se ve en la LLC. El LCM blástico leucémico puede imitar una leucemia aguda con células de tamaño intermedio a grande, aumento de la relación núcleo-citoplasma, cromatina finamente dispersa y nucleolo relativamente pequeño o inconspicuo. Varios estudios han reportado casos de LCM leucémico con células atípicas muy grandes y nucleolos prominentes imitando una leucemia prolinfocítica, que parecen corresponder a la fase leucémica de la variante pleomórfica del LCM. Estas células son más grandes que en la leucemia prolinfocítica y muestran marcadas irregularidades nucleares con contornos hiperlobados y cromatina moderadamente cohesiva. Algunos de estos casos tienen cariotipos hiperdiploides y se han asociado con la variante pleomórfica del LCM en el ganglio linfático.
Bazo
Macroscópicamente la afectación esplénica por LCM muestra un patrón micronodular generalizado, que se puede asociar ocasionalmente con infiltración perivascular. Los nódulos de la pulpa blanca están aumentados de tamaño, con una afectación variable de la pulpa roja (fig. 6A). Se pueden encontrar centros germinales residuales «desnudos» en el 50% de los casos. Las células tumorales muestran el mismo aspecto monótono que en otras localizaciones. Algunos casos pueden mostrar un patrón de zona marginal en la periferia de los nódulos, con células de citoplasma pálido y abundante.
![]()
Fig. 6: (A) Afectación esplénica e (B)
intestinal en el LCM.
Tracto gastrointestinal
Una manifestación común de la enfermedad gastrointestinal es la «poliposis linfomatoide», en la que se identifican múltiples pólipos linfoides en intestino delgado y grueso. Estos pólipos pueden estar asociados con grandes masas tumorales, habitualmente ileocecales y linfadenopatía regional (fig. 6B). Aunque esta presentación clinicopatológica es relativamente característica del LCM, también puede estar causada por otros linfomas no Hodgkin, particularmente el linfoma folicular y el linfoma de zona marginal tipo MALT.
En otros casos, los hallazgos macroscópicos consisten en úlceras superficiales, grandes masas tumorales o engrosamiento difuso de la mucosa. Un estudio prospectivo ha demostrado infiltración microscópica de la mucosa gastrointestinal por LCM, sin lesiones macroscópicas aparentes, en un elevado número de pacientes. En algunos casos la infiltración glandular por las células tumorales puede simular lesión linfoepitelial, haciendo difícil el diagnóstico diferencial con el linfoma de zona marginal. De todas formas la escasez de estas lesiones y el aspecto monótono del infiltrado linfoide debería sugerir el diagnóstico de LCM.
LCM compuesto y otros procesos linfoproliferativos
Recientemente se han descrito algunos casos de LCM asociados con un segundo linfoma maligno en la misma localización, LCM se ha encontrado con linfoma folicular, LLC, plasmocitoma y mieloma múltiple. Los estudios moleculares identificaron reordenamientos clonales independientes en estos tumores sugiriendo dos orígenes clonales distintos. Sin embargo, un caso mostró un reordenamiento clonal específico de IgH, común en el componente de linfoma folicular y en el de LCM y reordenamiento tanto de bcl-1 como de bcl-2 y expresión de CD5 y CD10 restringida a los componentes de LCM y linfoma folicular respectivamente. Estos hallazgos sugieren una rara evolución de una única clona maligna dando como resultado dos linfomas distintos morfológica, fenotípica y molecularmente.
Inmunofenotipo y hallazgos citogenéticos
El LCM es una proliferación de células B maduras que expresan los marcadores de línea B: CD19, CD20 (fig. 7A), CD22 y CD79a, e inmunoglobulinas de superficie con coexpresión de IgM e IgD y una tendencia a expresar cadenas ligeras lambda mayor que kappa. De forma similar a la LLC, el LCM coexpresa el antígeno de línea T, CD5, aunque ocasionalmente se pueden ver casos negativos (fig. 7B) (tabla 2). La detección inmunohistoquímica de CD23 suele ser negativa, aunque se han reportado raros casos blastoides positivos y en algunos casos se puede detectar débil positividad por citometría de flujo.
La detección inmunohistoquímica del inhibidor de kinasa dependiente de ciclina, p27, es útil en el diagnóstico diferencial, sobretodo cuando ciclina D1 es negativa (fig. 7D). En la mayoría de procesos linfoproliferativos p27 se expresa de forma inversa al índice proliferativo, pero en el LCM es independiente de éste, siendo habitualmente negativa en el LCM clásico y positiva en las variantes blastoides. La tricoleucemia también es negativa o débilmente positiva. La tinción de Ki-67 es útil para valorar la actividad proliferativa de estos tumores. En general la tinción con Ki-67 muestra pocas células positivas en la variante clásica, mientras que las variantes blastoides muestran un elevado número de células positivas.
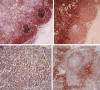
Fig. 7: Inmunofenotipo del linfoma de
células del manto. Las células tumorales expresan CD20 (A), con coexpresión de
CD5 (B) y sobreexpresión de ciclina-D1 (C). La expresión de p27 es baja en la
forma típica de LCM (D).
Ciclina D1
La sobreexpresión de ciclina D1 es un fenómeno constante y altamente específico del LCM , se asocia con el reordenamiento genético del locus de bcl-1 y se detecta en prácticamente todos los casos de LCM. La inmunoreactividad se detecta siempre en el núcleo de las células (fig. 7C), pero la intensidad puede variar de célula a célula y entre distintos casos, reflejando distintos parámetros, como los niveles de proteína y de mRNA, así como la interacción con otras proteínas. Ciclina D1 se detecta también en el núcleo de los histiocitos, de las células endoteliales y en las células epiteliales, que se pueden utilizar como control interno.
La técnica de detección inmunohistoquímica de ciclina D1 puede ser difícil. Se han desarrollado recientemente nuevas técnicas para detectar la sobreexpresión, incluyendo la reacción en cadena de la polimerasa transcriptasa reversa (RT-PCR), RT-PCR cuantitativa, hibridación in situ y citometría de flujo. Utilizando métodos sensibles ciclina D1 se detecta en prácticamente todos los casos de LCM. Además de en el LCM, ciclina D1 se sobreexpresa en un subgrupo de LLC, en algunos casos con t(11;14), leucemias prolinfocíticas y en el 25% de mielomas múltiples que tienen la translocación t(11;14) o amplificación del gen. Niveles bajos de ciclina D1 se pueden detectar en tricoleucemias sin reordenamiento de bcl-1.
La alteración citogenética característica del LCM es la translocación t(11;14)(q13;q32), aunque se han descrito ocasionalmente otras variantes afectando el punto de ruptura 11q13. Esta translocación se detecta con estudios citogenéticos clásicos en más del 65% de los LCM. Estudios citogenéticos convencionales, FISH e hibridación genómica comparativa (CGH) han revelado un elevado número de alteraciones cromosómicas secundarias en el LCM. Las alteraciones secundarias más comunes son las pérdidas de los cromosomas 1p, 6q, 8p, 9p, 10p, 11q, 13 y 17p y ganancias de 3q, 7p, 8q, 12q, 18q y Xq. Las variantes blastoides tienen cariotipos más complejos y niveles más elevados de amplificaciones de DNA que las variantes típicas. Además algunas alteraciones cromosómicas como la ganancia de 3q, 7p y 12q y pérdidas de 17p son significativamente más frecuentes en la variante blastoide que en la típica. La tetraploidía se encuentra más frecuentemente en la variante pleomórfica (80%) y en la blástica (36%) que en la típica (8%). Alteraciones del cromosoma 8q24, incluyendo la t(8;14)(q24;q32) y algunas variantes, se han identificado ocasionalmente en LCM blastoide con un curso clínico muy agresivo.
Diagnóstico diferencial
Algunos procesos reactivos que muestran folículos linfoides primarios expandidos o hiperplasia de la zona del manto pueden plantear el diagnóstico diferencial con el LCM de patrón nodular. Habitualmente en estos procesos benignos las células linfoides no muestran irregularidades nucleares, la arquitectura ganglionar está relativamente preservada y los pacientes suelen ser jóvenes y con adenopatía localizada. La negatividad para ciclina D1 y CD5, así como la ausencia de monoclonalidad descartan el diagnóstico de LCM.
Existen algunos linfomas B que muestran características morfológicas y fenotípicas de LCM pero en los que no se ha podido demostrar positividad para ciclina D1, no presentan la t(11;14), el contexto clínico es distinto y el pronóstico es significativamente mejor que en el LCM. Todo esto indica que debe tratarse de entidades distintas incluyendo LLC-B atípica, linfomas de zona marginal CD5 positivos o linfomas linfoplasmocitoides.
La afectación ganglionar por LLC puede tener un patrón predominantemente interfolicular e infiltrar la zona del manto, dando un aspecto de centros germinales sin manto aparente. El predominio de células pequeñas con núcleo redondo y la presencia de prolinfocitos y parainmunoblastos ayudará al diagnóstico de LLC, ya que estas características siempre están ausentes en el LCM. Las características fenotípicas pueden ayudar al diagnóstico, aunque puede existir superposición y deben interpretarse en el contexto morfológico y fenotípico.
Algunos casos de LCM tienen un marcado patrón nodular sugestivo de linfoma folicular. La monotonía de la población celular, con ausencia de centroblastos y las irregularidades nucleares deben sugerir el diagnóstico de LCM. Sin embargo pueden verse en el LCM ocasionales centroblastos de los centros germinales residuales, en estos casos las tinciones de inmunohistoquímica para CD5, ciclina D1, CD10 y bcl-6 pueden clarificar el diagnóstico.
En algunos casos de LCM la presencia de células tumorales con citoplasma pálido y abundante y centros germinales residuales, puede sugerir el diagnóstico de linfoma de la zona marginal. La identificación de áreas de LCM y la ausencia de una corona de células del manto rodeando los centros germinales reactivos debería hacer pensar en el diagnóstico de LCM. El inmunofenotipo y las características moleculares ayudaran al diagnóstico diferencial. Es importante reconocer estos tumores como LCM, pues el comportamiento clínico es muy agresivo.
El LCM pleomórfico se puede confundir con un linfoma B de célula grande. Las características nucleares del LCM pleomórfico con contornos hendidos e irregulares, cromatina dispersa y la relativa disociación entre el gran tamaño del núcleo y el pequeño nucleolo deberían sugerir el origen en las células del manto. La detección de CD5 y ciclina D1 facilita el diagnóstico, aunque existe un subgrupo de linfomas B de célula grande que muestran positividad para CD5, estos casos no tienen la translocación t(11;14) y son ciclina D1 negativos.
El LCM blástico se puede presentar como un proceso leucémico y las características citológicas de las células blásticas del manto pueden sugerir blastos linfoides o mieloides. Estas formas expresan el fenotipo típico del LCM y CD34 y TdT son negativos.
Existe un grupo de procesos linfoproliferativos leucémicos que muestran la translocación t(11;14) y características clínicas superponibles a las del LCM. Se ha sugerido que todos estos procesos deberían ser considerados variantes leucémicas del LCM. Sin embargo, algunos casos de LLC con t(11;14) muestran un curso clínico indolente, los pacientes con curso clínico agresivo están asociados con cariotipos complejos y por último análisis citogenéticos de neoplasias linfoides leucémicas con la t(11;14) están demostrando diferencias en las alteraciones cromosómicas, sugiriendo que no se trate de procesos uniformes. Etiquetar bajo el diagnóstico de LCM todos los procesos linfoproliferativos leucémicos con t(11;14) podría obstaculizar la comprensión de la historia natural y las diferencias biológicas de procesos posiblemente diferentes.
Curso clínico y factores pronósticos
La evolución clínica de los pacientes con LCM es relativamente agresiva con respuesta pobre al tratamiento. La supervivencia total media es de alrededor de 3-4 años. La remisión completa se obtiene en el 6-35% de los pacientes y el periodo de supervivencia libre de enfermedad es corto. Aunque la presentación clínica inicial de las variantes blastoides es similar a la del LCM típico, la evolución clínica es mucho más agresiva y habitualmente no obtienen remisiones completas.
Diversos estudios han mostrado que la morfología blastoide se asocia a mal pronóstico, pero las variantes blastoides habitualmente van asociadas a otros parámetros de mal pronóstico. El parámetro pronóstico con significado adverso más consistentemente reportado es el índice mitótico elevado (más de 1.5-2.5 mitosis por campo de gran aumento). En estudios multivariantes la citología blastoide pierde su significado, mientras que el índice mitótico se mantiene como factor independiente. El papel del patrón de zona del manto como indicador de buen pronóstico es controvertido. Estudios de FISH y CGH han demostrado una evolución clínica más agresiva en pacientes con cariotipos complejos. Los LCM que tienen inactivación concomitante de las vías de p16INK4a y ARF/ p53, tienen un comportamiento más agresivo que los que muestran solamente mutaciones de p53 o sin alteraciones en ninguno de éstos genes. Los factores clínicos asociados a peor pronóstico son la edad avanzada, el mal estado general, estadio avanzado, esplenomegalia, LDH elevada, niveles bajos de albúmina sérica, enfermedad tumoral y anemia.
BIBLIOGRAFÍA
Jaffe ES, Harris NL, Stein H, VardimanJW. World Health Organisation classification of tumors. Pathology and genetics of tumors of haematopoietic and lymphoid tissues. 2001.
Campo E, Raffeld M, Jaffe ES. Mantle-cell lymphoma. Semin Hematol 1999; 36: 115-27.
Bosch F, Lopez-Guillermo A, Campo E, et al. Mantle cell lymphoma: presenting features, response to therapy, and prognostic factors. Cancer 1998; 82: 567-75.
Campo E, Raffeld M, Jaffe ES. Mantle-cell lymphoma. Semin Hematol 1999; 36: 115-27.
Molina TJ, Delmer A, Cymbalista F, et al. Mantle cell lymphoma, in leukaemic phase with prominent splenomegaly. A report of eight cases with similar clinical presentation and aggressive outcome. Virchows Arch 2000; 437: 591-8.
Montserrat E, Bosch F, Lopez-Guillermo A, et al. CNS involvement in mantle-cell lymphoma. J Clin Oncol 1996; 14: 941-4.
Singleton TP, Anderson MM, Ross CW, Schnitzer B. Leukemic phase of mantle cell lymphoma, blastoid variant. Am J Clin Pathol 1999; 111: 495-500.
Bertoni F, Zucca E, Cotter FE. Molecular basis of mantle cell lymphoma. Br J Haematol 2004; 124: 130-40.
Su W, Yeong KF, Spencer J. Immunohistochemical analysis of human CD5 positive B cells: mantle cells and mantle cell lymphoma are not equivalent in terms of CD5 expression. J Clin Pathol 2000; 53: 395-7.
Hummel M, Tamaru J, Kalvelage B, Stein H. Mantle cell (previously centrocytic) lymphomas express VH genes with no or very little somatic mutations like the physiologic cells of the follicle mantle. Blood 1994; 84: 403-7.
Camacho FI, Algara P, Rodriguez A, et al. Molecular heterogeneity in MCL defined by the use of specific VH genes and the frequency of somatic mutations. Blood 2003; 101: 4042-6.
Du MQ, Diss TC, Xu CF, et al. Ongoing immunoglobulin gene mutations in mantle cell lymphomas. Br J Haematol 1997; 96: 124-31.
Kienle D, Krober A, Katzenberger T, et al. VH mutation status and VDJ rearrangement structure in mantle cell lymphoma: correlation with genomic aberrations, clinical characteristics, and outcome. Blood 2003; 102: 3003-9.
Pinyol M, Campo E, Nadal A, et al. Detection of the bcl-1 rearrangement at the major translocation cluster in frozen and paraffin-embedded tissues of mantle cell lymphomas by polymerase chain reaction. Am J Clin Pathol 1996; 105: 532-7.
Athanasiou E, Kotoula V, Hytiroglou P, et al. In situ hybridization and reverse transcription-polymerase chain reaction for cyclin D1 mRNA in the diagnosis of mantle cell lymphoma in paraffin-embedded tissues. Mod Pathol 2001; 14: 62-71.
Swerdlow SH, Zukerberg LR, Yang WI, Harris NL, Williams ME. The morphologic spectrum of non-Hodgkin’s lymphomas with BCL1/cyclin D1 gene rearrangements. Am J Surg Pathol 1996; 20: 627-40.
Bosch F, Campo E, Jares P, et al. Increased expression of the PRAD-1/CCND1 gene in hairy cell leukaemia. Br J Haematol 1995; 91: 1025-30.
Zukerberg LR, Benedict WF, Arnold A, et al. Expression of the retinoblastoma protein in low-grade B-cell lymphoma: relationship to cyclin D1. Blood 1996; 88: 268-76.
Jares P, Campo E, Pinyol M, et al. Expression of retinoblastoma gene product (pRb) in mantle cell lymphomas. Correlation with cyclin D1 (PRAD1/CCND1) mRNA levels and proliferative activity. Am J Pathol 1996; 148: 1591-600.
Pinyol M, Hernandez L, Cazorla M, et al. Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood 1997; 89: 272-80.
Pinyol M, Cobo F, Bea S, et al. p16(INK4a) gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin’s lymphomas. Blood 1998; 91: 2977-84.
Pinyol M, Hernandez L, Martinez A, et al. INK4a/ARF locus alterations in human non-Hodgkin’s lymphomas mainly occur in tumors with wild-type p53 gene. Am J Pathol 2000; 156: 1987-96.
Dreyling MH, Bullinger L, Ott G, et al. Alterations of the cyclin D1/p16-pRB pathway in mantle cell lymphoma. Cancer Res 1997; 57: 4608-14.
Hernandez L, Fest T, Cazorla M, et al. p53 gene mutations and protein overexpression are associated with aggressive variants of mantle cell lymphomas. Blood 1996; 87: 3351-9.
Chiarle R, Budel LM, Skolnik J, et al. Increased proteasome degradation of cyclin-dependent kinase inhibitor p27 is associated with a decreased overall survival in mantle cell lymphoma. Blood 2000; 95: 619-26.
Quintanilla-Martinez L, Thieblemont C, Fend F, et al. Mantle cell lymphomas lack expression of p27Kip1, a cyclin-dependent kinase inhibitor. Am J Pathol 1998; 153: 175-82.
Stilgenbauer S, Winkler D, Ott G, et al. Molecular characterization of 11q deletions points to a pathogenic role of the ATM gene in mantle cell lymphoma. Blood 1999; 94: 3262-4.
Traweek ST, Sheibani K, Winberg CD, et al. Monocytoid B-cell lymphoma: its evolution and relationship to other low-grade B-cell neoplasms. Blood 1989; 73: 573-8.
Cohen PL, Kurtin PJ, Donovan KA, Hanson CA. Bone marrow and peripheral blood involvement in mantle cell lymphoma. Br J Haematol 1998; 101: 302-10.
Onciu M, Schlette E, Medeiros LJ, et al. Cytogenetic findings in mantle cell lymphoma cases with a high level of peripheral blood involvement have a distinct pattern of abnormalities. Am J Clin Pathol 2001; 116: 886-92.
Wong KF, Chan JK, So JC, Yu PH. Mantle cell lymphoma in leukemic phase: characterization of its broad cytologic spectrum with emphasis on the importance of distinction from other chronic lymphoproliferative disorders. Cancer 1999; 86: 850-7.
Piris MA, Mollejo M, Campo E, et al. A marginal zone pattern may be found in different varieties of non-Hodgkin’s lymphoma: the morphology and immunohistology of splenic involvement by B-cell lymphomas simulating splenic marginal zone lymphoma. Histopathology 1998; 33: 230-9.
Isaacson PG, MacLennan KA, Subbuswamy SG. Multiple lymphomatous polyposis of the gastrointestinal tract. Histopathology 1984; 8: 641-56.
Romaguera JE, Medeiros LJ, Hagemeister FB, et al. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma. Cancer 2003; 97: 586-91.
Romaguera JE, Medeiros LJ, Hagemeister FB, et al. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma. Cancer 2003; 97: 586-91.
Cachia AR, Diss TC, Isaacson PG. Composite mantle-cell lymphoma and plasmacytoma. Hum Pathol 1997; 28: 1291-5.
Fend F, Quintanilla-Martinez L, Kumar S, et al. Composite low grade B-cell lymphomas with two immunophenotypically distinct cell populations are true biclonal lymphomas. A molecular analysis using laser capture microdissection. Am J Pathol 1999; 154: 1857-66.
Tsang P, Pan L, Cesarman E, Tepler J, Knowles DM. A distinctive composite lymphoma consisting of clonally related mantle cell lymphoma and follicle center cell lymphoma. Hum Pathol 1999; 30: 988-92.
Kumar S, Green GA, Teruya-Feldstein J, Raffeld M, Jaffe ES. Use of CD23 (BU38) on paraffin sections in the diagnosis of small lymphocytic lymphoma and mantle cell lymphoma. Mod Pathol 1996; 9: 925-9.
Quintanilla-Martinez L, Thieblemont C, Fend F, et al. Mantle cell lymphomas lack expression of p27Kip1, a cyclin-dependent kinase inhibitor. Am J Pathol 1998; 153: 175-82.
de Boer CJ, van Krieken JH, Schuuring E, Kluin PM. Bcl-1/cyclin D1 in malignant lymphoma. Ann Oncol 1997; 8 Suppl 2: 109-17.
de Boer CJ, van Krieken JH, Kluin-Nelemans HC, Kluin PM, Schuuring E. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995; 10: 1833-40.
Chan JK, Miller KD, Munson P, Isaacson PG. Immunostaining for cyclin D1 and the diagnosis of mantle cell lymphoma: is there a reliable method? Histopathology 1999; 34: 266-70.
Miranda RN, Briggs RC, Kinney MC, et al. Immunohistochemical detection of cyclin D1 using optimized conditions is highly specific for mantle cell lymphoma and hairy cell leukemia. Mod Pathol 2000; 13: 1308-14.
Athanasiou E, Kotoula V, Hytiroglou P, et al. In situ hybridization and reverse transcription-polymerase chain reaction for cyclin D1 mRNA in the diagnosis of mantle cell lymphoma in paraffin-embedded tissues. Mod Pathol 2001; 14: 62-71.
Pruneri G, Fabris S, Baldini L, et al. Immunohistochemical analysis of cyclin D1 shows deregulated expression in multiple myeloma with the t(11;14). Am J Pathol 2000; 156: 1505-13.
Swerdlow SH, Zukerberg LR, Yang WI, Harris NL, Williams ME. The morphologic spectrum of non-Hodgkin’s lymphomas with BCL1/cyclin D1 gene rearrangements. Am J Surg Pathol 1996; 20: 627-40.
Wlodarska I, Pittaluga S, Hagemeijer A, Wolf-Peeters C, Van Den BH. Secondary chromosome changes in mantle cell lymphoma. Haematologica 1999; 84: 594-9.
Onciu M, Schlette E, Medeiros LJ, et al. Cytogenetic findings in mantle cell lymphoma cases with a high level of peripheral blood involvement have a distinct pattern of abnormalities. Am J Clin Pathol 2001; 116: 886-92.
Ott G, Kalla J, Ott MM, et al. Blastoid variants of mantle cell lymphoma: frequent bcl-1 rearrangements at the major translocation cluster region and tetraploid chromosome clones. Blood 1997; 89: 1421-9.
Hunt JP, Chan JA, Samoszuk M, et al. Hyperplasia of mantle/marginal zone B cells with clear cytoplasm in peripheral lymph nodes. A clinicopathologic study of 35 cases. Am J Clin Pathol 2001; 116: 550-9.
Vasef MA, Medeiros LJ, Koo C, McCourty A, Brynes RK. Cyclin D1 immunohistochemical staining is useful in distinguishing mantle cell lymphoma from other low-grade B-cell neoplasms in bone marrow. Am J Clin Pathol 1997; 108: 302-7.
Yamaguchi M, Seto M, Okamoto M, et al. De novo CD5+ diffuse large B-cell lymphoma: a clinicopathologic study of 109 patients. Blood 2002; 99: 815-21.
Bosch F, Lopez-Guillermo A, Campo E, et al. Mantle cell lymphoma: presenting features, response to therapy, and prognostic factors. Cancer 1998; 82: 567-75.
Majlis A, Pugh WC, Rodriguez MA, Benedict WF, Cabanillas F. Mantle cell lymphoma: correlation of clinical outcome and biologic features with three histologic variants. J Clin Oncol 1997; 15: 1664-71.
![]()