
Vol. 36, n.º 4, 2003
|
REVISTA
ESPAÑOLA DE
Vol. 36, n.º 4, 2003 |
José Cameselle Teijeiro1, Manuel Sobrinho-Simoes2
1 Departamento de Anatomía
Patológica, Hospital Clínico Universitario, Universidad de Santiago de
Compostela, Santiago de Compostela, España, apjocame@usc.es
2 Departamento de Patología, Facultad de
Medicina de Oporto e Instituto de Patología Molecular e Inmunología de la
Universidad de Oporto (IPATIMUP), Oporto, Portugal, sobrinho.simoes@ipatimup.pt
RESUMEN
Debido al uso generalizado de la citología por punción-aspiración con aguja fina, cada vez es más frecuente encontrar nódulos tiroideos con algunas características morfológicas de carcinoma papilar cuyo diagnóstico es un problema difícil. En este artículo se revisan los criterios diagnósticos de este tipo histológico de carcinoma bien diferenciado. Se discute el diagnóstico diferencial con el adenoma folicular con hiperplasia papilar y se abordan aspectos controvertidos en relación con el diagnóstico de la variante folicular, con énfasis en las formas no invasivas. También se consideran la variante sólida, la variante cribiforme-morular –incluyendo el carcinoma tiroideo asociado a poliposis adenomatosa familiar y algunas neoplasias relacionadas tales como los tumores trabeculares hialinizantes y los tumores híbridos.
Palabras clave: carcinoma papilar, variante folicular, variante cribiforme-morular, variante sólida, tumor híbrido, tumor trabecular hialinizante.
SUMMARY
Due to a more widespread use of fine-needle aspiration cytology it is becoming more and more frequent to find thyroid nodules with some morphological features of papillary carcinoma that are difficult to diagnose. Here we review the diagnostic criteria of this histological type of well-differentiated carcinoma. We discuss the differential diagnoses considering the difficulties occasionally raised by follicular adenoma with papillary hyperplasia, as well as the controversial aspects related with the encapsulated follicular variant of papillary carcinoma. We also review the solid variant and the cribriform-morular variant –including thyroid carcinoma associated to familial adenomatous polyposis. This discussion is followed by a brief description of some closely related neoplasms such as hybrid tumours and hyalinizing trabecular tumours.
Key words: papillary carcinoma, follicular variant, cribriform-morular variant, solid variant, hybrid tumours, hyalinizing trabecular tumours.
INTRODUCCIÓN
El carcinoma papilar (CP) de la glándula tiroides ha sido definido por la OMS como un «tumor maligno epitelial que evidencia diferenciación de célula folicular, típicamente con estructuras papilares y foliculares así como cambios nucleares característicos (aspecto esmerilado, pálido y/o vacío, tamaño grande, contorno irregular, hendiduras profundas, nucleolo pequeño y pseudoinclusiones)» (1,2). La clave para su diagnóstico la constituyen las características nucleares, mientras que la presencia de invasión vascular o capsular, no constituye un requisito necesario (1-5). Las características nucleares se han convertido en el equivalente de las papilas en el diagnóstico del CP y los tumores que en el pasado se designaban como «carcinoma mixto papilar-folicular» deben ser reclasificados como CP (2).
El verdadero adenoma papilar (adenoma folicular con hiperplasia papilar o variante papilar de adenoma folicular) es un tumor benigno encapsulado y parcialmente quístico que aparece fundamentalmente en niños y adolescentes (4,6,7). Histológicamente muestra papilas y folículos pero los núcleos son redondos y de situación basal y por definición las características nucleares del CP están ausentes (fig. 1). También suele acompañarse de depósitos de hemosiderina en el citoplasma del epitelio folicular.

Fig. 1. Adenoma papilar (adenoma
folicular con hiperplasia papilar). Como se aprecia en el recuadro, los núcleos
carecen de las características del carcinoma papilar.
Aunque si se exceptúa la variante folicular difusa (o multinodular) de CP (8), el diagnóstico de las formas invasivas del CP no suele representar un problema, existe en la actualidad el temor a pasar por alto un tumor maligno y una tendencia a «sobrediagnosticar» la variante folicular de carcinoma papilar y sobre todo las formas bien delimitadas y encapsuladas (Tumor de Lindsay) (4,9). Chan (10) ha insistido recientemente en la necesidad de aplicar criterios estrictos a la hora de diagnosticar la variante folicular encapsulada del CP debido a que aún en el caso de equivocarse y diagnosticar como adenoma a esta variante de CP, la simple excisión de la lesión es ya curativa, mientras que un falso diagnóstico de malignidad dará lugar a una tiroidectomía, a tratamiento con I-131 y a un trauma psicológico innecesario. Sin embargo, debido a las presiones legales y de endocrinólogos y cirujanos, cada vez con mayor frecuencia, los extendidos procedentes de la punción aspiración con aguja fina (PAAF) de lesiones tiroideas con datos citológicos mínimos o focales de PC se diagnostican como atípicos o sospechosos; por lo que es más frecuente encontrar nódulos tiroideos con algunas características morfológicas de CP, de manera focal o difusa, pero no totalmente establecidas, que constituyen un dificultad diagnóstica seria. Aunque menos del 1% de los enfermos con la variante folicular encapsulada de CP fallecen a causa del tumor, más del 25% tienen metástasis a ganglios linfáticos regionales (10) y esta variante puede presentarse en ocasiones como una enfermedad metastásica de primario oculto (11). También se han descrito metástasis en la variante macrofolicular de CP, la cual tiene un patrón arquitectural que imita a un nódulo hiperplásico o a un adenoma de tipo macrofolicular (12). Por ello el patólogo tampoco puede elevar el umbral de los requisitos diagnósticos y dejar de diagnosticar estas lesiones.
En un intento por solucionar el problema Renshaw y Gould (13) propusieron la realización de estudios que documentasen la variabilidad entre observadores expertos al diagnosticar estas lesiones, los cuales podrían ser utilizados como un argumento defensivo en el caso de una segunda opinión discrepante. Uno de estos estudios acaba de ser publicado y confirmó tanto la variabilidad en el diagnóstico realizado por diferentes expertos en patología tiroidea, como la dificultad para definir de manera precisa el tipo de aclaramiento nuclear diagnóstico de CP (14). Otra propuesta de Renshaw y Gould fue la de una conferencia de consenso para intentar definir los criterios mínimos para el diagnóstico de la variante folicular de CP (13). En un enfoque práctico, el grupo de patólogos de Chernobil (15) han propuesto las categorías de tumor bien diferenciado de potencial maligno incierto para designar aquellos tumores encapsulados que tienen sólo mínimos cambios nucleares de los que se ven en el CP típico, y de carcinoma bien diferenciado, sin otra especificación para aquellos tumores con invasión capsular y/o vascular que tengan también cambios nucleares no concluyentes de CP.
En este trabajo se evalúan algunos problemas relacionados con el diagnóstico de del CP, con énfasis en las variantes de patrón folicular. Se revisan también algunos subtipos de CP descritos recientemente y los tumores de tipo trabecular hialinizante, los cuales son considerados por algunos investigadores como una forma de CP.
VARIANTES FOLICULARES DE CARCINOMA PAPILAR
Con independencia del pragmatismo de Williams et al (15) el patólogo debe intentar en la medida de lo posible el diagnóstico de la variante folicular del CP (fig. 2), a causa del diferente comportamiento clínico, biológico y las distintas alteraciones moleculares que caracterizan al CP en comparación con el carcinoma folicular. Así por ejemplo, mientras que los reordenamientos RET/PTC aparecen en cerca del 65% de los CP de la población general (16-18), los reordenamientos PAX8-PPARg se detectan en cerca del 55% de los carcinomas foliculares y 10% de los adenomas foliculares, pero no se observaron en los CP ni en los tumores de células de Hürthle (19,20). Además, el CP es más frecuente en pacientes con historia de exposición a radiación ionizante en los cuales el porcentaje de reordenamientos RET/PTC es superior al 87% (16). Por otra parte, existe una asociación entre radiación, reordenamiento RET/PTC3 y la variante sólida de CP (21,22), así como entre el reordenamiento RET/PTC1 y los carcinomas de pequeño tamaño con patrón de crecimiento papilar (fenotipo «bonsai») (23). Recientemente se ha sugerido que la mutación BRAF(V599E) está asociada al carcinoma papilar como un evento etiopatogénico alternativo al reordenamiento RET/PTC (24,25).

Fig. 2. Variante folicular de
carcinoma papilar.
Baloch y LiVolsi (26) han propuesto la designación de tumores híbridos para algunos de estos tumores que combinan características clinicopatológicas de carcinoma folicular y de CP. Aunque no se puede excluir la existencia de verdaderos carcinomas híbridos, en nuestra opinión esta terminología es desafortunada ya que las vías moleculares de transformación de las neoplasias foliculares y del CP son diferentes, y la situación se corresponde más con nuestra incapacidad para clasificar a estas lesiones que una bona fide neoplasia híbrida. Desde el punto de vista práctico, la terminología propuesta por grupo de Chernobil (15) parece suficiente para clasificar estas lesiones mientras que otros estudios no aporten nueva información (27).
Desafortunadamente, el análisis molecular no puede ser utilizado como ayuda para el diagnóstico de la variante folicular del CP debido a que la traslocación RET/PTC: a) ocurre en aproximadamente dos tercios de los CP (16,17), b) se ha encontrado, aunque por un solo grupo de investigadores, en tumores benignos y malignos de células de Hürthle con independencia de las características nucleares (28), y c) también se ha descrito en procesos no neoplásicos (18,29). La utilización de lectinas (30) o de marcadores inmunohistoquímicos tales como citoqueratinas de alto peso molecular, citoqueratina 19, vimentina, involucrina, HBME1, CD57 (Leu 7), CD15 (LeuM1), CD44, S100, antígenos relacionados con los grupos sanguíneos y de galectinas, que se expresan con mayor frecuencia en los CP, tampoco es por ahora efectiva en el diagnóstico diferencial de casos concretos debido a que incluso en CP típicos su expresión puede ser focal o negativa. (10,31-35). Tampoco el estudio inmunohistoquímico de la expresión del PPARg ha resultado útil para distinguir las neoplasias foliculares de la variante folicular de carcinoma papilar (35). Por ello, el diagnóstico del CP es un todavía puramente morfológico y por tanto subjetivo. Para hacer el diagnóstico de la variante folicular de CP, los cambios nucleares deben ser generalizados y no focales, los núcleos deben ser claros, vacíos, con hendiduras, amontonados por la pérdida de la polaridad alrededor del folículo y preferiblemente con alguna pseudoinclusión. La presencia de folículos alargados, coloide oscuro, papilas abortivas o bien formadas, cuerpos de psammoma, histiocitos multinucleados y focos de infiltración favorecen el diagnóstico de CP. El reconocimiento de las características nucleares del CP exige excluir el aclaramiento artefactual, que por defecto de fijación, se produce en la porción central algunas neoplasias foliculares y el artefacto de burbuja, también por fijación inadecuada, que simula una pseudoinclusión pero carece de verdadera membrana nuclear delimitante (4). La comparación de las características nucleares de las células neoplásicas con las del tejido tiroideo adyacente suelen ayudar a la valoración y mientras que la transición brusca sugiere carcinoma, la transición gradual indica benignidad. En raras ocasiones puede aparecer un CP en un adenoma folicular como una masa que crece bien delimitada de las áreas no atípicas (3,4). Sin embargo, no existe ningún dato morfológico aislado que sea patognomónico de CP, por lo que Chan (10) ha propuesto una serie de criterios «mayores» y «menores» que permitirían el diagnóstico de la variante folicular encapsulada de CP (tabla 1).

La variante macrofolicular de CP ha sido definida como una neoplasia encapsulada cuyas células tienen características nucleares de CP y un patrón de crecimiento macrofolicular que ocupa más del 50% del área tumoral. Debido a su apariencia microscópica, esta forma de CP, de pronóstico excelente, puede ser confundida un nódulo hiperplásico con macrofolículos rellenos de coloide (12).
La variante folicular difusa (o multinodular) ha sido caracterizada como una forma invasiva de CP que afecta masivamente a un lóbulo o a toda la glándula (8,36,37). Ocurre casi exclusivamente en mujeres jóvenes, con un patrón de crecimiento difuso o multinodular que simula clínica y macroscópicamente un bocio multinodular hiperplásico. Histológicamente, los nódulos tienen un borde de crecimiento expansivo y generalmente carecen de cápsula bien definida. Aunque no hay una buena delimitación entre el tumor y el parénquima adyacente, las células tienen características nucleares típicas de CP clásico (fig. 3). La mayoría de los nódulos están constituidos por microfolículos o trabéculas y también se observan folículos de tamaño medio y macrofolículos. Se trata de una forma agresiva de CP que muestra invasión vascular en el 80% de los casos, metástasis ganglionar también en el 80% y extensión extraglandular en el 70% de los enfermos. En esta variante se ha encontrado una mayor inmunoexpresión del receptor-activador del plasminógeno tipo-uroquinasa (uPA-R) y Sialyl Lewis X que podría estar relacionada con la invasividad de este subtipo tumoral (8).
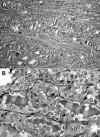
Fig. 3. A) Variante folicular difusa
de carcinoma papilar. B) Como puede apreciarse a mayor aumento, las
características nucleares son típicas de carcinoma papilar.
ADENOMA TRABECULAR HIALINIZANTE Y CARCINOMA PAPILAR
El adenoma trabecular hialinizante (ATH) es una neoplasia de células foliculares cuyos núcleos son indistinguibles de los del CP (fig. 4A). Es una forma infrecuente de adenoma que tiene un patrón de crecimiento característico de tipo trabecular y/o «zellballen» en un estroma hialinizado que simula amiloide (38,39). Las células son alargadas y se disponen perpendiculares al eje mayor de la trabécula con los núcleos a diferentes niveles. Su apariencia histológica peculiar hace necesario el estudio inmunohistoquímico para su diferenciación del paraganglioma y de algunos carcinomas medulares. El ATH es positivo para tiroglobulina y aunque ocasionalmente hay coexpresión de somatostatina y/o neurotensina, no se ha observado immunorreactividad para calcitonina, péptido del gen relacionado con la calcitonina ni para cromogranina (40). Desde su descripción en 1987, se ha descrito su contrapartida maligna, el carcinoma trabecular hialinizante, con capacidad para la invasión local y las metástasis, por lo que ambas neoplasias se han incluido ahora en el grupo denominado tumores trabeculares hialinizantes (TTHs) (40,41).

Fig. 4. Adenoma trabecular
hialinizante. B) Intensa inmunorreactividad de membrana y citoplasmática para
MIB-1.
Los TTHs comparten con el CP numerosas características morfológicas que incluyen tanto las rasgos nucleares como la presencia de cuerpos de psammoma (38-40). Se han descrito casos de CP y tumor trabecular hialinizante coexistiendo en la misma glándula tiroides (42,43), de CP con áreas de tipo ATH (43) y casos de microcarcinoma papilar en el seno de ATH (41). Muchos TTTs ocurren en glándulas con tiroiditis de Hashimoto, un antecedente frecuente también en el CP (38,39,42-44). También se ha descrito la existencia de un patrón de crecimiento de tipo ATH focal en la variante cribiforme-morular de CP (45). Por todo ello y dado que los TTHs y los carcinomas papilares comparten también un patrón anómalo de distribución intra y extracelular del material de la membrana basal (46) y el perfil de inmunoexpresión para citoqueratinas (47,48) y galectina-3 (34), diversos investigadores han propuesto que se trata de dos tipos de neoplasias relacionadas y que los TTHs pueden representar una variante de CP (46-48). Otros investigadores (49,50), en base a la presencia de cuerpos amarillos intracitoplasmaticos, y su exclusiva (dentro de la glándula tiroides) inmunopositividad citoplasmática y de membrana para el anticuerpo MIB-1(fig. 4B), apoyan la idea de que el ATH es una entidad clinicopatológica diferente del CP. Por otra parte, la descripción más reciente de reordenamientos RET/PTC, considerados específicos del CP, en TTHs ha sido presentada como una prueba de que estos tumores representan una variante morfológica de CP (44,51). Sin embargo, debido a que estos reordenamentos también se han descrito en condiciones no neoplásicas tales como la tiroiditis de Hashimoto (18,27), la controversia se mantiene. Nosotros no hemos encontrado mutaciones del gen APC al examinar una serie de 8 TTHs (52). El carcinoma trabecular hialinizante descrito en una paciente con poliposis adenomatosa familiar (39) y recogido en la serie de Cetta et al (53) representa, en nuestra opinión, un CP de la variante cribiforme-morular. Con independencia de si los TTHs son una forma de CP, no se ha descrito ningún ATH que se haya comportado de forma maligna (54) por lo que parece razonable considerar a los TTHs como una categoría histológica y diferenciar las formas benignas (adenoma trabecular hialinizante) de las malignas (carcinoma trabecular hialinizante), en base a los criterios tradicionales aplicables a las otras neoplasias foliculares (54).
VARIANTE SÓLIDA DE CARCINOMA PAPILAR
La variante sólida ha sido propuesta como una forma de CP dotada de una ligera mayor frecuencia de metástasis a distancia y un pronóstico menos favorable que el CP clásico (55). Esta variante ha sido caracterizada como una neoplasia que tiene un patrón de crecimiento sólido/trabecular/insular en >70% del nódulo tumoral primario, conserva las características citológicas de CP y no muestra focos de necrosis (55). Debido a que la variante sólida de CP tiene un pronóstico sólo ligeramente menos favorable que el CP clásico, con una tasa de supervivencia a los diez años del 90%, es muy importante distinguirla de los carcinomas tiroideos pobremente diferenciados (56). A diferencia del carcinoma pobremente diferenciado la variante sólida de CP conserva las características nucleares del CP clásico, no muestra focos de necrosis, tiene mínima o nula actividad mitósica, y carece de mutaciones de p53.
VARIANTE CRIBIFORME-MORULAR DE CARCINOMA PAPILAR
Aunque el gen APC no está implicado en la tumorigénesis de los carcinomas papilares habituales de la glándula tiroides (57,58), los pacientes con poliposis adenomatosa familiar (FAP) tienen una predisposición a sufrir tumores tiroideos, además de pólipos intestinales y otras alteraciones (59-61). En 1994, Harach et al (62) caracterizaron el carcinoma tiroideo que aparece en los pacientes con FAP como un tumor peculiar, con características histológicas específicas, lo que ha sido confirmado por otros grupos (55-57). Otros investigadores confirmaron que se trata de una forma de CP (60,63). Por otra parte, también se ha descrito la existencia de casos esporádicos de carcinomas tiroideos indistinguibles histológicamente de los que aparecen en pacientes con FAP, que han sido denominados variante cribiforme-morular de CP (45,64). Esta variante se caracteriza histológicamente por un patrón de crecimiento complejo en el que se entremezclan áreas de tipo cribiforme, folicular, papilar, trabecular y sólido con mórulas de apariencia escamoide (fig. 5A). Las áreas trabeculares son reminiscentes del ATH (fig. 5B) y las células de las áreas morulares tienen núcleos ricos en biotina con un peculiar aclaramiento de la cromatina. Las células tumorales son cuboidales o altas, con pseudoestratificación nuclear, citoplasma abundante eosinófilo a oxifílico y núcleos hipercromáticos o indistinguibles del CP típico. En la punción-aspiración, las extensiones de esta variante muestran, además de las características citológicas típicas del CP clásico, grupos y placas arremolinadas de células fusiformes con amplio citoplasma, dispuestas en un patrón papilar, folicular y sólido (64,65). La existencia de este subtipo cribiforme-morular como una forma esporádica del carcinoma tiroideo asociado a FAP y su relación con mutación somática del gen APC ha sido confirmada recientemente (64). En estos tumores también se han encontrado mutaciones del gen de la b-catenina así como alteraciones la expresión de su proteína (66). La variante cribiforme-morular predomina en mujeres jóvenes, pero mientras que en su forma esporádica se presenta como un nódulo bien delimitado, los casos asociados a FAP son a menudo multicéntricos debido a diferentes mutaciones somáticas añadidas a la mutación germinal (60). A causa de que su comportamiento biológico es similar al del CP clásico, es importante distinguir esta variante de otras formas agresivas de carcinoma tiroideo como la variante de células altas del CP, el carcinoma de células columnares, o el carcinoma pobremente diferenciado (45,64).
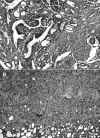
Fig. 5. A) Variante cribiforme-morular
de carcinoma papilar. B) En las áreas de patrón de crecimiento trabecular esta
neoplasia es indistinguible del adenoma trabecular hialinizante.
AGRADECIMIENTOS
Trabajo realizado con ayuda de la Xunta de Galicia (Proyecto n.º PGIDT03PXI91801PR).
BIBLIOGRAFÍA
Hedinger C, Williams ED, Sobin LH, editors. Histological typing of thyroid tumors. WHO International Histological Classification of Tumors, 2nd ed. Berlin: Springer-Verlag; 1988.
Hedinger C, Williams ED, Sobin LH. He WHO histological classification of thyroid tumors: a commentary on the second edition. Cancer 1989; 63: 908-11.
Rosai J, Carcangiu ML, DeLellis RA. Tumors of the thyroid gland. En: Rosai J, editor. Atlas of Tumor Pathology. Third Series, Fascicle 5. Washington, DC: Armed Forces Institute of Pathology; 1992.
Chan JKC. Tumors of the thyroid and parathyroid glands. En: Fletcher CDM, editor. Diagnostic Histopathology of Tumors. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2000. p. 959-1056.
Baloch ZW, LiVolsi VA. Pathology of thyroid gland. En: LiVolsi VA, Asa SL, eds. Endocrine Pathology. 1st ed. Philadelphia, Pennsylvania: Churchill Livingstone; 2002. p. 61-101.
Vuitch F, Leavitt A. Papillary variant of follicular adenoma of thyroid [abstract]. Mod Pathol 1990; 3: 104A.
Mai KT, Landry DC, Thomas J, Burns BF, Commons AS, Yazdi HM, Odell PF. Follicular adenoma with papillary architecture: a lesion mimicking papillary thyroid carcinoma. Histopathology 2001; 39: 25-32.
Ivanova R, Soares P, Castro P, Sobrinho-Simoes M. Diffuse (or multinodular) follicular variant of papillary carcinoma: a clinicipathologic and immunohistochemical analysis of ten cases of an aggressive form of differentiated thyroid carcinoma. Virchows Arch 2002; 440: 418-24.
Renshaw AA, Gould EW. Why there is the tendency to «overdiagnose» the follicular variant of papillary thyroid carcinoma. Am J Clin Pathol 2002; 117: 19-21.
Chan JKC. Strict criteria should be applied in the diagnosis of encapsulated follicular variant of papillary thyroid carcinoma. Am J Clin Pathol 2002; 117: 16-8.
Baloch ZW, LiVolsi VA. Encapsulated follicular variant of papillary thyroid carcinoma with bone metastases. Mod Pathol 2000; 13: 861-5.
Albores-Saavedra J, Gould E, Vardaman C, Vuitch F. The macrofollicular variant of papillary thyroid carcinoma: a study of 17 cases. Hum Pathol 1991; 22: 1195-205.
Renshaw AA, Gould EW. Why there is the tendency to «overdiagnose» the follicular variant of papillary thyroid carcinoma. Am J Clin Pathol 2002; 117: 19-21.
Hirokawa M, Carney JA, Goellner JR, DeLellis RA, Heffess CS, Katoh R, et al. Observer variation of encapsulated follicular lesions of the thyroid gland. Am J Surg Pathol 2002; 26: 1508-14.
Williams ED, Abrosimov A, Bogdanova T, Ito M, Rosai J, Sidirov Y, et al. Guests editorial: two proposals regarding the terminology of thyroid tumors. Int J Surg Pathol 2000; 8: 181-3.
Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol 2002; 13: 3-16.
Lam KY, Lo CY, Leung PS. High prevalence of RET proto-oncogene activation (RET/PTC) in papillary thyroid carcinomas. Eur J Endocrinol 2002; 147: 741-5.
Sheils OM, Oéary JJ, Uhlmann V, Lattich K, Sweeney EC. Ret/PTC-1 activation in Hashimoto tiroiditis. Int J Surg Pathol 2000; 8: 185-9.
Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARg rearrangement in thyroid tumors. RT-PCR and immunohistochemical analyses. Am J Surg Pathol 2002; 26: 1016-23.
Marques AR, Espadinha C, Catarino AL, Moniz S, Pereira T, Sobrinho LG, et al. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab 2002; 87: 3947-52.
Nikirov YE, Rowland JM, Bove KE, Monforte-Muñoz H, Fagin JA. Distinct pattern of RET oncogene rearrangements in morphologic variants of radiation-induced and sporadic thyroid carcinomas in children. Cancer Res 1997; 57: 1690-4.
Thomas GA, Bunnell H, Cook HA, Williams ED, Nerovnya A, Cherstvoy ED, et al. High prevalence of RET/PTC rearrangements in Ukrainian and Belarussian post-Chernobil thyroid papillary carcinomas: a strong correlation between RET/PTC3 and the solid-follicular variant. J Clin Endocrinol Metab 1999; 84: 4232-8.
Soares P, Fonseca E, Wynford-Thomas D, Sobrinho-Simoes M. Sporadic ret - rearranged papillary carcinoma of the thyroid: a subset of slow growing, less aggressive thyroid neoplasms? J Pathol 1998; 185: 71-8.
Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, et al BRAF mutation and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003; 22: 4578-80.
Nikiforova MN, Kimura ET, Gandhi M, Bidinger PW, Knauf JA, Basolo F, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab 2003; 88: 5399-404.
Baloch ZW, LiVolsi VA. Follicular-patterned lesions of the thyroid. The bane of the pathologst. Am J Clin Pathol 2002; 117: 143-50.
Castro P, Fonseca E, Magalhaes J, Sobrinho-Simoes M. Follicular, papillary, and «hybrid» carcinomas. Endocr Pathol 2002; 13: 313-20.
Chiappetta G, Toti P, Cetta, Giuliano A, Pentimalli F, Amendola I, et al. The RET/PTC oncogene is frequently activated in oncocytic thyrid tumors (Hürthle cell adenomas and carcinomas), but not in oncocytic hyperplastic lesions. J Clin Endocrinol Metab 2002; 87: 364-9.
Arif S, Blanes A, Díaz-Cano SJ. Hashimoto’s thyroiditis shares features with early papillary thyroid carcinoma. Histopathology 2002; 41: 357-62.
Sobrinho-Simoes M, Damjanov I. Lectin histochemistry of papillary and follicular carcinoma of the thyroid gland. Arch Pathol Lab Med 1986; 110: 722-9.
Jennings TA, Boguniewicz AB, Sheehan CE, Figge J. Involucrin selectively stains papillary thyroid carcinoma. Appl Imunohistochem 1998; 6: 55-61.
Alves P, Soares P, Fonseca E, Sobrinho-Simoes M. Papillary thyroid carcinoma overexpresses fully and underglycosylated mucins together with native and sialylated mucin antigens and histo-blood group antigens. Endocr Pathol 1999; 10: 315-24.
Rorive S, Eddafali B, Fernández S, Decaestecker C, André S, Kaltner H, et al. Changes in galectin-7 and cytokeratin-19 expression during the progression of malignancy in thyroid tumors: diagnostic and biological implications. Mod Pathol 2002; 15: 1294-301.
Gaffney RL, Carney TJ, Sebo TJ, Erickson LA, Volante M, Papotti M, et al. Galectin-3 expression in hyalinizing trabecular tumors of the thyroid. Am J Surg Pathol 2003; 27: 494-8.
Feilchenfeldt J, Tötsch M, Sheu S-Y, Robert J, Spiliopoulos A, Frilling A. Expression of galectin-3 in normal and malignant thyroid tissue by quantitative PCR and immunohistochemistry. Mod Pathol 2003; 16: 1117-23.
Sobrinho-Simoes M, Soares J, Carneiro F, Limbert E. Diffuse follicular variant of papillary carcinoma of the thyroid: report of eight cases of a distinct aggressive type of thyroid tumor. Surg Pathol 1995; 3: 189-203.
Mizukami Y, Nonomura A, Michigishi T, Ohmura K, Noguchi M, Ishizaki T. Diffuse follicular variant of papillary carcinoma of the thyroid. Histopathology 1995; 27: 575-7.
Carney JA, Goellner JR. Hyalinizing trabecular adenoma of the thyroid gland. Am J Surg Pathol 1987; 11: 583-91.
Cameselle JM, Sambade C, Varela J, Villanueva JP, Varela R, Sobrinho-Simoes. Adenoma trabecular hialinizante de tiroides. Estudio inmunocitoquímico y revisión. Rev Esp Patol 1990; 23: 215-20.
Sambade C, Franssila KO, Cameselle-Teijeiro J, Nesland J, Sobrinho-Simoes M. Hyalinizing trabecular adenoma. A misnomer for a peculiar tumor of the thyroid gland. Endocr Pathol 1991; 2: 83-91.
Molberg K, Albores-Saavedra J. Hyalinizing trabecular carcinoma of the thyroid gland. Hum Pathol 1994; 25: 192-7.
Chetty R, Beydoun R, LiVolsi VA. Paraganglioma-like (hyalinizing trabecular) adenoma of the thyroid revisited. Pathology 1994; 26: 429-31.
Li M, Rosai J, Carcangiu ML. Hyalinizing trabecular adenoma of the thyroid. A distinct tumor or a pattern of growth? Evaluation of 28 cases [abstract]. Lab Invest 1995; 72: 54A.
Cheung CC, Boerner SL, MacMillan CM, Ramyar L, Asa SL. Hyalinizing trabecular tumor of the thyroid: a variant of papillary carcinoma proved by molecular genetics. Am J Surg Pathol 2000; 24: 1622-6.
Cameselle-Teijeiro J, Chan JKC. Cribriform-morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma? Mod Pathol 1999; 12: 400-11.
Li M, Carcangiu ML, Rosai J. Abnormal intracellular and extracellular distribution of basement membrane material in papillary carcinoma and hyalinizing trabecular tumors ofthe thyroid: implication for deregulation of secretory pathways. Hum Pathol 1997: 28: 1366-72.
Fonseca E, Nesland JM, Sobrinho-Simoes M. Expression of stratified epithelial-type cytokeratins in hyalinizing trabecular adenomas supports their relationship with papillary carcinomas of the thyroid. Histopathology 1997; 31: 330-5.
Papotti M, Riella P, Montemurro F, Pietriebasi F, Bussolati G. Immunophenotypic heterogeneity of hyalinizing trabecular tumors of the thyroid. Histopathology 1997; 31: 525-33.
Rothenberg HJ, Goellner JR, Carney JA. Hyalinizing trabecular adenoma of the thyroid: recognition and characterization of its cytoplasmic yellow body. Am J Surg Pathol 1999, 23: 118-25.
Hirokawa M, Carney JA. Cell membrane and cytoplasmic staining for MIB-1 in hyalinizing trabecular adenoma of the thyroid gland. Am J Surg Pathol 2000; 24: 575-8.
Papotti M, Volante M, Giuliano A, Fassina A, Fusco A, Bussolati G, et al. RET/PTC activation in hyalinizing trabecular tumors of the thyroid. Am J Surg Pathol 2000; 24: 1615-21.
Cameselle-Teijeiro J, Ruíz-Ponte C, Reyes-Santías RM, Cameselle-Teijeiro JF, Alfonsín-Barreiro N, Sobrinho-Simoes M. Evidence against involvement of APC gene mutation in hyalinizing trabecular tumors. Pathologica 2002; 94: 74-5.
Cetta F, Montalto G, Gori M, Curia C, Cama A, Olschwang S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: results from a European cooperative study. J Clin Endocrinol Metab 2000; 85: 286-92.
LiVolsi VA. Hyalinizing trabecular tumor of the thyroid: adenoma, carcinoma, or neoplasm of uncertain malignant potential? Am J Surg Pathol 2000; 24: 1683-4.
Nikiforov YE, Erickson LA, Nikiforova MN, Caudill CM, Lloyd RV. Solid variant of papillary thyroid carcinoma. Incidence, clinical-pathologic characteristics, molecular analysis, and biologic behavior. Am J Surg Pathol 2001; 25: 1478-84.
Sobrinho-Simoes M, Sambade C, Fonseca E, Soares P. Poorly differentiated carcinomas of the thyroid: a review of the clinicopathologic features of a series of 28 cases of heterogeneous, clinically aggressive group of thyroid tumours. Int J Surg Pathol 2002; 10: 123-31.
Colletta G, Sciacchitano S, Palmirotta R, Ranieri A, Zanella E, Cama A, et al. Analysis of adenomatous polyposis coli gene in thyroid tumors. Br J Cancer 1994; 70: 1985-8.
Curtis L, Wyllie AH, Shaw JJ, Williams GT, Radulescu A, DeMicco C, et al. Evidence against involvement of APC mutation in papillary thyroid carcinoma. Eur J Cancer 1994; 30: 984-7.
Soravia C, Sugg SL, Berk T, Mitri A, Cheng H, Gallinger S, et al. Familial adenomatous poliposis-associated thyroid cancer: a clinical, pathological, and molecular genetics study. Am J Pathol 1999; 154: 127-35.
Miyaki M, Iijima T, Ishii R, Hishima T, Mori T, Yoshinaga K, et al. Molecular evidence for multicentric development of thyroid carcinomas in patients with familial adenomatous polyposis. Am J Pathol 2000; 157: 1825-7.
Iwama T, Konishi M, Iijima T, Yoshinaga K, Tominaga T, Koike M, et al. Somatic mutation of the APC gene in thyroid carcinoma associated with familial adenomatous polyposis. Jpn J Cancer Res 1999; 90: 372-6.
Harach HR, Williams GT, Williams DE. Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular neoplasm. Histopathology 1994; 25: 549-61.
Cetta F, Toti P, Petracci M, Montalto G, Disanto A, Lore F, et al. Thyroid carcinoma associated with familial adenomatous polyposis. Histopathology 1997; 31: 231-6.
Cameselle-Teijeiro J, Ruiz-Ponte C, Loidi L, Suárez-Peñaranda JM, Baltar J, Sobrinho-Simoes M. Somatic but not germline mutation of the APC gene in a case of cribriform-morular variant of papillary thyroid carcinoma. Am J Clin Pathol 2001; 115: 486-93.
Chong J-M, Koshiishi N, Kurihara K, Kubono S, Kawai T, Fukayama M. Aspiration and imprint cytopathology of thyroid carcinoma associated with familial adenomatous polyposis. Diagn Cytopathol 2000; 23: 101-5.
Xu B, Yoshimoto K, Miyaguchi A, Kuma S, Mizusawa N, Hirokawa M, et al. Cribriform-morular variant of papillary thyroid carcinoma: a pathological and molecular genetics study with evidence of frequent somatic mutations in exon 3 of the b-catenin gene. J Pathol 2003; 199: 58-67.
![]()