
Vol. 36, n.º 1, 2003
|
REVISTA
ESPAÑOLA DE
Vol. 36, n.º 1, 2003 |
J. M. Viguer1, B. Vicandi1, P. López Ferrer1, J. A. Jiménez-Heffernan2
1 Sección de Citología. Dpto. Anatomía Patológica. Hospital Universitario La Paz. 2 Servicio de Anatomía Patológica. Hospital General y Universitario de Guadalajara.
INTRODUCCIÓN
La PAAF es un procedimiento diagnóstico que se utiliza de forma rutinaria en el estudio de las adenomegalias desde finales de los años setenta (1). En nuestro país el primer trabajo en ese sentido fue una comunicación conjunta, con una casuística de algo menos de 1000 casos, del Hospital Virgen del Rocío de Sevilla, Clínico de Zaragoza y Doce de Octubre y La Paz de Madrid, presentada en el Congreso Europeo de Citología que se celebró en Paris en 1983.
Desde entonces, debido al entusiasmo de algunos y a la «bondad de la técnica», el procedimiento se ha extendido de forma muy considerable. En Diciembre de 2001, el Hospital La Paz contaba con una casuística de 6.861 casos de PAAF de ganglio linfático. Este desarrollo se ha producido de forma paralela en todo el mundo, hecho que se pone de manifiesto por la gran cantidad de trabajos sobre esta materia que encontramos en la literatura (2,3,4).
Por otra parte, el desarrollo del arsenal de técnicas complementarias, como la inmunohistoquímica y la biología molecular, aplicables en el material de PAAF (5,6,7), han aportado más solidez y capacidad al diagnóstico de la patología linfática mediante el uso de la citología.
La PAAF en el protocolo diagnóstico de las adenopatías debe situarse en el lugar adecuado, es decir en el primer escalón. En ese lugar, la punción no solo aporta su buena sensibilidad, especificidad y predictibilidad, sino también una gran capacidad de racionalizar recursos, ya que no es lo mismo la actitud frente a una linfadenitis inespecífica que frente a un ganglio con metástasis o linfoma (8).
Así entendido y con una correcta indicación de la técnica, el patrón citológico que encontraremos con más frecuencia es el de linfadenopatías no neoplásicas y de estas, las linfadenitis reactivas inespecíficas, seguidas de cerca por las metástasis y con mucha menos frecuencia por los linfomas.
Dentro de los casos que podemos incluir en el grupo de linfadenitis no neoplásicas existen algunos procesos, bien conocidos desde el punto de vista histopatológico, de los que encontramos pocas referencias de sus patrones citológicos y que podríamos clasificarlos en: Linfadenopatías con histiocitos peculiares, linfadenopatías eosinofílicas y linfadenopatías en pacientes con alteraciones de la inmunidad.
1. LINFADENOPATíAS CON HISTIOCITOS PECULIARES
Dentro de este grupo podemos incluir la histiocitosis sinusal con linfadenopatía masiva (Enfermedad de Rossai-Dorfman), la linfadenitis de Kikuchi, la granulomatosis de células de Langerhans y la linfadenitis por toxoplasma (Linfadenitis de Piringer-Kuchinka).
a) Histiocitosis sinusal con linfadenopatía masiva (Enfermedad de Rossai-Dorfman)
Clínicamente se caracteriza por la presencia de adenomegalias cervicales bilaterales con fiebre, leucocitosis, aumento de la velocidad de sedimentación globular e hipergammaglobulinemia policlonal. Afecta sobre todo a jóvenes y si bien su localización típica es la linfática, puede afectar a otros órganos tales como órbita, piel, SNC, tracto respiratorio superior e incluso tracto digestivo y genitourinario. Su etiología es desconocida, aunque podría corresponder a un proceso infeccioso.
Desde el punto de vista histológico se caracteriza por una marcada dilatación de los senos ganglionares, en cuyo interior junto a linfocitos y células plasmáticas se identifican histiocitos con intensa linfofagocitosis (emperipolesis).
Citológicamente se caracteriza por la presencia de frotis celulares con patrón reactivo, en los que destacan la presencia de histiocitos grandes, de citoplasma delicado que se disponen predominantemente aislados pero que en ocasiones constituyen pequeños grupos. Estas células pueden contener en sus citoplasmas eritrocitos, polimorfonucleares y células plasmáticas. Sin embargo, el hallazgo clave es la emperipolesis (9,10), es decir la presencia de linfocitos aparentemente dentro de los citoplasmas histiocitarios (fig. 1). Con técnica de Papanicolaou puede observarse un halo claro de membrana histiocitaria, alrededor de los linfocitos fagocitados (11), (fig. 2). La realización de estudio inmunihistoquímico sobre el material de PAAF revelará positividad para proteina s-100.
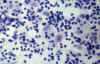
Fig. 1. Sobre un fondo linfocitario se observan histiocitos de citoplasma
amplio con fenómenos de linfofagocitosis. PAP 40x.
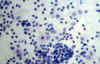
Fig. 2. Linfofagocitosis. Se
observa un halo claro, bien definido. PAP 40x.
Estos hallazgos, junto a la ausencia de auténticos granulomas y el estudio inmuhistoquímico, permiten reconocer esta enfermedad en el material de PAAF.
El diagnóstico diferencial debe plantearse con otras enfermedades con histiocitos peculiares, enfermedades granulomatosas e incluso procesos malignos ya que en ocasiones los histiocitos presentan núcleos voluminosos y nucléolos prominentes (fig. 3). En ocasiones, en linfadenitis inespecíficas, microfragmentos de centros germinales que asocian histiocitos y linfocitos pueden remedar fenómenos de emperipolesis.
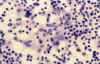
Fig. 3. Grupo de histiocitos en el
que se observa algún núcleo voluminosos con nucleolo prominente. PAP 40x.
b) Linfadenitis necrotizante (Linfadenitis de Kikuchi)
Es una enfermedad autolimitada que clínicamente se caracteriza por adenopatías cervicales, de tamaño mediano, asociadas a fiebre. Presenta cierta predilección por el sexo femenino y por la raza asiática.
Su etiología es desconocida aunque podría tener relación tanto con procesos infecciosos frente a algunos virus tales como Adenovirus, Parvovirus y virus del grupo Herpes como el tipo 6, tipo 8, Ebstein Barr y Citomegalovirus, como con reacciones autoinmunes.
Histológicamente presenta focos de necrosis con intensa cariorrexis en la paracortical que pueden confluir afectando a gran parte del ganglio. Estos focos se acompañan de inmunoblastos y macrófagos. Se describen tres subtipos: Tipo proliferativo, tipo necrotizante y tipo xantomatoso. En realidad estos tipos no son más que diferentes patrones en diferente momento evolutivo de la enfermedad.
Desde el punto de vista citológico los frotis en general presentan alta celularidad, con patrón polimorfo reactivo y presencia de macrófagos con cuerpos tingibles.
Los hallazgos claves son la presencia de importante cariorrexis, con numerosos restos nucleares sueltos por el frotis (50% de nuestros casos) y la existencia de histiocitos «pequeños». Estos últimos presentan dos morfologías características: Con núcleo en media luna y restos nucleares fagocitados (fig. 4), o con núcleo muy convolucionado (12,13,14), (fig. 5).
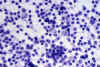
Fig. 4. Histiocitos con núcleo en
media luna y fagocitosis PAP 40x.

Fig. 5. Histiocitos pequeños con
núcleo convolucionado. Diff-Quick 40x.
El diagnóstico diferencial varía según la fase proliferativa, necrotizante o xantomatosa. En ocasiones, en las formas necrotizantes, el material de PAAF puede presentarse con escasa celularidad y abundante material necrótico granular (fig. 6) que recuerda a las linfadenitis caseificadas. En esos casos, la ausencia de granulomas y de polimorfonucleares, pueden ser de ayuda para reconocer esta entidad. Por otra parte, dado que la afectación no es difusa, podemos obtener un cuadro citológico de linfadenitis inespecífica. Si la fase proliferativa es muy exuberante puede plantear problemas de diagnóstico diferencial con linfomas. La imagen es idéntica a la afectación en el ganglio linfático por lupus eritematoso diseminado. Este diagnostico diferencial debe basarse en hallazgos clínicos y analíticos.
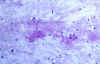
Fig. 6. Abundante material
necrótico en un frotis hipocelular. PAP 20x.
c) Granulomatosis de células de Langerhans
Se caracteriza por la proliferación de células de Langerhans que son un tipo especial (dendrítico) de histiocitos.
Este enfermedad se considera como un proceso reactivo peculiar frente a un agente desconocido. Desde el punto de vista clínico incluye el Síndrome de Letterer-Sive, la enfermedad de Hans Schüller-Christian y el granuloma eosinófilo, con diferente afectación de órganos y pronóstico. La afectación ganglionar por este proceso puede darse en el contexto de una afectación sistémica o como única manifestación.
Desde el punto de vista histológico, en la afectación ganglionar por esta patología se observa la infiltración de los senos por células de Langerhans junto con eosinófilos y células multinucleadas. La célula de Langerhans se caracteriza por ser una célula de aspecto histiocitario, con citoplasma denso eosinófilo y núcleo con indentaciones y pliegues (grano de café).
Citologicamente se observa un frotis celular que contiene eosinofilos, polinucleares y células gigantes (fig. 7), junto con células de Langerhans de citoplasma denso no fagocítico y núcleo característico (15,16,17), (fig. 8). Imunohistoquimicamente, estas células presentan positividad para proteina S-100 y CD 1a . El estudio ultrastructural del material de PAAF presentará gránulos de Birbeck.
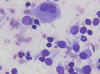
Fig. 7. Histiocito multinucleado
junto con eosinófilos y células de Langerhans. Dic-Quick 40x.
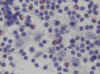
Fig. 8. Células de Langerhans, con
núcleo con hendiduras. PAP 40x.
El diagnóstico diferencial debe plantearse con adenopatias con células gigantes, linfadenitis eosinofílicas y otras linfadenitis con histiocitos peculiares. No obstante la imagen citológica es bastante característica y con la ayuda de técnicas complementarias, es posible reconocer esta enfermedad en el material de PAAF.
d) Toxoplasmosis (Linfadenitis de Piringer-Kuchinka)
Clínicamente se caracteriza por la presencia de adenopatias cervicales de larga evolución en pacientes jóvenes.
Histológicamente se observa intensa hiperplasia folicular, pequeños acúmulos de celulas epiteliodes sin necrosis y presencia de linfocitos B monocitoides en los senos.
Desde el punto de vista citológico, la clave se encuentra en la presencia de microgranulomas no necrotizantes, en el contexto de una linfadenitis reactiva. Estos microgranulomas estan constituidos por pocas células de aspecto histiocitario, de citoplasmas poligonales o panzudos y núcleos redondos u ovales relativamente grandes (18,19), (fig. 9). En algunas ocasiones es posible identificar al agente causal en el material de PAAF (20).
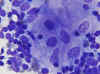
Fig. 9. Grupo de histiocitos de
citoplasma amplio y núcleo ovoide. Dic-Quick 40x.
El diagnostico diferencial debe establecerse sobre todo con granulomas tuberculoides. El pequeño tamaño de los acúmulos histiocitarios, el borde en general bien definido de los mismos, la ausencia o escasa cantidad de células epiteliodes alargadas y la ausencia de necrosis, pueden facilitar el reconocimiento de esta entidad.
2. LINFADENOPATíAS CON EOSINÓFILOS
En este grupo se incluyen procesos reactivos a drogas, parásitos y la enfermedad de Kimura que es a la que nos vamos a referir.
Enfermedad de Kimura
Es una enfermedad poco frecuente, de etiología desconocida que se manifiesta por masa en la región de cabeza y cuello con adenopatías. Las adenopatías pueden ser la única manifestación de la enfermedad que en ocasiones puede afectar a glándulas salivales.
Los pacientes presentan además eosinofília en sangre periférica y elevación de la IgE en suero. En algunos casos se acompaña de síndrome nefrótico.
Histologicamente se caracteriza por hiperplasia folicular con proliferación vascular y densa infiltración por eosinófilos que pueden constituir abscesos. También pueden verse células gigantes tipo Warthin-Finkeldey.
Citologicamente el hallazgo mas característico es la presencia de gran cantidad de eosinófilos, con o sin células gigantes, en un fondo linfocitario reactivo (21,22,23). Si bien la imagen citológica no es característica, en un contexto clínico-analítico concordante, la PAAF puede orientar el diagnóstico que debe ser confirmado por biopsia. Sin embargo, en un paciente diagnosticado de enfermedad de Kimura, la PAAF puede hacer innecesarias biopsias repetidas.
El diagnóstico diferencial debe establecerse con todas las adenopatias con eosinófilos, desde la enfermedad de Hodgkin a la granulomatosis de células de Langerhans.
3. LINFADENOPATíAS EN PACIENTES CON ALTERACIONES DE LA INMUNIDAD
En este apartado nos vamos a referir a dos de estas situaciones: La infección por el virus de la inmunodeficiencia humana VIH, en su fase aguda (Síndrome de linfadenopatía persistente generalizada) y a los síndromes linfoproliferativos posttransplante.
a) Infección por VIH
La infección por VIH causa una alteración grave del sistema inmune que desemboca en el Síndrome de Inmunodeficiencia Adquirido (SIDA). La seroconversión de un paciente se acompaña generalmente de un síndrome febril con adenopatías que remeda al cuadro clínico de la mononucleosis infecciosa.
Este cuadro agudo se acompaña de viremia, con difusión del virus a los tejidos linfoides, seguida de una respuesta inmune y de un periodo de latencia que puede prolongarse durante años (24).
La utilidad de la PAAF en estos pacientes se fundamenta en que en muchos casos puede obviarse la biopsia quirúrgica, ya que la citología frecuentemente identifica las infecciones específicas y la malignidad. La utilización simúltanea de estudios microbiológicos y de biología molecular mejoran los resultados.
Clínicamente el síndrome de linfadenopatía persistente generalizada se define como la presencia de adenopatías en dos o más localizaciones (excluyendo la inguinal ), de más de un centímetro, con una persistencia de más de tres meses.
Histologicamente se caracteriza por un proceso evolutivo en el que se pueden distinguir tres fases: Hiperplasia folicular, involución folicular y deplección linfoide (24).
Desde el punto de vista citológico el cuadro no es específico, observándose en la fase de hiperplasia folicular un cuadro de linfadenitis reactiva con gran actividad de centros germinales (fig. 10). Las otras dos fases presentan frotis hipocelulares inespecíficos (8).
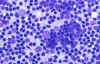
Fig. 10. Frotis muy celular con
abundantes células de los centros germinales. Diff-Quick 40x.
Cuando se instaura el síndrome de inmunodeficiencia adquirida, los cuadros citológicos mas frecuentes son las infecciones por micobacterias, leishmanias (25,25,27) y hongos.
b) Síndromes linfoproliferativos posttransplante
Los síndromes linfoproliferativos posttransplante son un conjunto heterogéneo de proliferaciones linfoides, asociados con mucha frecuencia al virus Ebstein-Barr que afectan a menos del 5% de los pacientes transplantados (28,29). Reconocer estos cuadros tiene mucho interés ya que mejoran considerablemente disminuyendo la supresión.
Histológicamente, la Organización Mundial de la Salud (30) clasifica estas lesiones en cuatro grandes categorías: 1) Lesiones iniciales, con dos formas, la hiperplasia reactiva plasmocítica y el cuadro similar a la mononucleosis infecciosa. 2) Síndrome linfoproliferativo posttransplante polimorfo con una forma policlonal y otra monoclonal.3)Síndrome linfoproliferativo posttransplante monomorfo, que se clasifica de acuerdo a la clasificación de los linfomas. De estos los más frecuentes son los linfomas B de células grandes, los tipo Burkitt y los linfomas T periféricos. 4) Otros tipos menos frecuentes como el parecido a la enfermedad de Hodgkin y plasmocitomas.
Citologicamente, excluyendo los linfomas que no se discuten en esta revisión, el cuadro es poco específico. Se observa un cuadro polimorfo con linfocitos grandes «atípicos» (fig. 11) y diferenciación plasmocitoide (31). En el momento actual, la citología es más útil en estos pacientes para descartar infecciones específicas o linfomas que para el diagnostico propiamente dicho de estas patologías.

Fig. 11. Patrón polimorfo reactivo
con abundantes linfocitos grandes. Dic-Quick 40x.
BIBLIOGRAFÍA
Lopes Cardozo P. The significance of fine needle aspiration cytology for the diagnosis and treatement of malignant lymphomas. Folia Haematol ( Leipz ) 1980; 107(4): 601-20.
Kocjan G. The role of FNAC in the diagnosis of lymph node enlargements. Cytopathology 1997; 8 (supple ): 2-3.
Thomas JO, Adeyi A, Amanguno H. Fine-needle aspiration in the manegement of peripheral lymphadenopaty in a developing country. Diagn Cytopathol 1999; 21(3): 159-62.
Steel BL, Schwartz MR, Ramzy I. Fine needle aspiration biopsy in the diagnosis of lymphadenopaty in 1.103 patients. Role, limitations and analysis of diagnostic pitfalls. Acta Cytol 1995; 39(1): 76-81.
Tani EM, Christensson B, Porwit A, Skoog L. Inmunocytochemical analysis and cytomorphologic diagnosis on fine needle aspirates of lymphoproliferatives disorders. Acta Cytol 1988; 32(2): 209-15.
Dunphy CH, Ramos R. Combining fine needle aspiration and flow cytometric inmunophenotyping and evaluation of nodal and extranodal sites for possible lymphoma: a retrospective review. Diagn Cytopathol 1997; 16(3): 200-6.
Grosso LE, Collins BT. DNA polymerase chain reaction using fine needle aspiration biopsy smears to evaluate non-Hodgkin’s lymphoma. Acta Cytol 1999; 43(5):837-41.
Kocjan G, ed. Clinical Cytopathology of the Head and Neck. London: Greenwich Medical Media, 2001.
Layfield LJ. Fine needle aspiration cytology findings in a case of sinus histiocytosis with massive lymphadenopaty (Rosai-Dorfman syndrome). Acta Cytol 1990; 34(6): 767-70.
Alvarez Alegret R, Martinez Tello A, Ramirez T, Gallego P, Martinez D, Garcia Julian G. Sinus histiocytosis with massive lymphadenopaty (Rosai- Dotfman disease): diagnosis with fine needle aspiration in a case with nodal and nasal involvement. Diagn Cytopathol; 13(4): 333-5.
Viguer JM, Jimenez-Heffernan JA, Lopez Ferrer P, Vicandi B. Importance of Papanicolaou-stained smears and inmunocytochemistry in the diagnosis of Rosai-Dorfman disease (letter). Acta Cytol 1999; 43(2): 328-30.
Tsang WY, Chan JK,. Fine needle aspiration cytology of Kikuchi’s Lymphadenitis. A report of 27 cases. Am J Clin Pathol 1994; 102(4): 454-8.
Tong TR, Chan OW, Lee K. Diagnosing Kikuchi disease on fine needle aspiration biopsy. A retrospective study of 44 cases diagnosed by cytology and 8 cases by histopathology. Acta Cytol 2001; 45(6): 953-7.
Viguer JM, Jimenez-Heffernan JA, Perez P, Lopez Ferrer P, Gonzalez Peramato P, Vicandi B. Fine needle aspiration Cytology of Kikuchi’s lymphadenitis: a report of ten cases. Diagn Cytopathol 2001; 25(4): 220-4.
Lee JS, Lee MC, Park CS, Juhng SW. Fine needle aspiration cytology of Langerhans cell histiocytosis confined to lymph nodes. A case report . Acta Cytol 1997; 41(6): 1793-6.
Akhtar M, Ali MA, Bakry M, Sackey K. Fine needle aspiration biopsy of Langerhans histiocytosis (Histiocytosis-X). Diagn Cytopathol 1993; 9(5): 527-33.
Kumar PV, Mousavi A, Karimi M, Bedayat GR. Fine needle aspiration of Langerhans cell histiocytosis of the lymph nodes. A report of six cases. Acta Cytol 2002; 46(4): 753-6.
Christ ML, Feltes-Kennedy M. Fine needle aspiration cytology of toxoplasmic lymphadenitis. Acta Cytol 1982; 26(4): 425-8.
Macey-Dare LV, Kocjan G, Goodman JR. Acquired toxoplasmosis of a submandibular lymph node in a 9-year-old boy diagnosed by fine needle aspiration cytology. Int J.Paediatr Dent 1996; 6(4): 265-9.
Zaharopoulos P. Demostration of parasites in toxoplasma lymphadenitis by fine needle aspiration cytology: a report of two cases. Diagn Cytopathol 2000; 22(1): 11-5.
Jarayama G, Peth KB. Fine needle aspiration Cytology in Kimura’s disease. Diagn Cytopathol 1995; 13(4): 295-9.
Chow LT, Yuen RW, Tsui WM, Ma TK, Chow WH, Chan SK. Cytologic features of Kimura’s disease in fine needle aspirates. A study of eigth cases. Am J Clin Pathol 1994; 102(3): 316-21.
Kini V, Shariff S. Cytodiagnosis of Kimura’s disease. Indian J Pathol Microbiol 1998;41(4): 473-7.
Strauchen JA, ed. Diagnostic histopathology of the lymph node New York: Oxford University Press, 1998.
Vicandi B, Jimenez-Heffernan JA, Lopez Ferrer P, Ortega L, Viguer JM. Cytologic diagnosis of leishmaniasis in HIV infecction. A report of eigth cases. Acta Cytol 2000; 44(5): 835-9.
Perez Guillermo M, Hernandez Gil A, Bonmatí C. Diagnosis of cutaneous leishmaniasis by fine needle aspiration cytology. Report a case. Acta Cytol 1998; 32(4): 485-88.
Tallada N, Raventós A, Martinez S, Compañó C, Almirante B. Leishmania lymphadenitis diagnosed by fine needle aspiration biopsy. Diagn Cytopathol 1993; 9(6): 673-6.
Gattuso P, Castelli MJ, Peng Y, Reddy BV. Posttransplant lymphoproliferative disorders. A fine needle aspiration biopsy study. Diagn Cytopathol 1997; 16(5): 392-5.
Dusenbery D, Nalesnik MA, Locker J, Swerdlow SH. Cytologic features of post-transplant lymphoproliferative disorders. Diagn Cytopathol 1997; 16(6): 489-96.
Jaff ES, Harris NL, Stein H, Vardiman JW. Ed. World Health Organozation Classification of Tumours. Pathology and genetic of tumours of haematopoietic and lymphoid tissues. IARC Press. Lyon 2001.
Davey DD, Gulley ML, Walker WD, Zaleski J. Cytologic findings in posttransplant lymphoproliferative disorders. A fine needle aspiration biopsy study. Acta Cytol 1990; 34(3): 304-310.
![]()