
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
Isabel García González, Miguel Valenzuela Serrano
Servicio de Anatomía Patológica. Hospital Regional Universitario Carlos Haya. Málaga.
INTRODUCCIÓN
Hoy en día es bien conocido que un amplio número de enfermedades glomerulares se caracterizan por presentar depósitos microfibrilares o microtubulares en microscopia electrónica. La amiloidosis es el prototipo de estas glomerulopa tías. Otras enfermedades sistémicas como crioglobulinemia, disproteinemias o LES también pueden tener depósitos organizados en el riñón. Recientemente han sido descritos dos procesos primarios no amiloidóticos con este tipo de alteración ultraestructural denominados «glomerulonefritis fibrilar» caracterizada por fibrillas que miden aproximadamente 20 nm de diámetro y «glomerulopatía inmunotactoide» tipificada por depósitos más grandes microtubulares. La posible relación entre estos dos procesos, a veces agrupados bajo el término de «glomerulopatía fibrilar inmunotactoide» y su potencial asociación con otras enfermedades sistémicas es materia de debate.
Cuando se analiza la literatura se advierte considerable falta de acuerdo en cuanto se refiere a la clasificación de las glomerulopatías con depósitos organizados debido en parte a su relativa rareza y heterogeneidad clínica siendo importante distinguir entre las diferentes entidades ya que el diagnóstico tiene implicaciones terapéuticas y pronósticas.
CLASIFICACIÓN DE LAS ENFERMEDADES CON DEPÓSITOS FIBRILARES GLOMERULARES
La demostración ultraestructural de depósitos organizados no es patognomónica de ninguna entidad clínica. Korbet y col. (1) han propuesto, para este tipo de enfermedades glomerulares, un esquema de clasificación basado en la presencia o ausencia de parámetros clínicos y serológicos suplementados con histoquímica (rojo Congo) e inmunohistoquímica (inmunoglobulinas) en la biopsia renal (tabla I). El primer paso es descartar amiloidosis que puede ser confirmada o excluida con la tinción de rojo Congo. Seguidamente, estudios de inmunofluorescencia valorarán si los depósitos no congófilos son derivados o no de inmunoglobulinas. Cuando los depósitos fibrilares no contienen inmunoglobulinas pueden estar relacionados con diabetes mellitus o contener componentes estructurales de la matriz tales como colágeno III o fibronectina. Si los depósitos contienen inmunoglobulinas se evaluarán datos clínicos y de laboratorio, que pueden llevar al reconocimiento de enfermedades sistémicas o linfoproliferativas subyacentes, de forma que según el criterio de Korbet y cols. cuando no hubiera evidencia de crioglobulinemia, LES o una paraproteina en suero u orina, podría hacerse el diagnóstico de glomerulopatía inmunotactoide. Otros investigadores hacen una subdivisión posterior en «glomerulonefritis fibrilar» y «glomerulopatía inmunotactoide» basados en el diámetro y organización fibrilar (2).

A continuación se comentarán detalladamente las glomerulopatías fibrilar e inmunotactoide y posteriormente se hará un breve comentario de otras enfermedades con depósitos glomerulares organizados limitado a la apariencia ultraestructural de los mismos y formas de diagnóstico diferencial.
GLOMERULOPATÍA FIBRILAR E INMUNOTACTOIDE
Rosenmann y Eliakin (3) fueron los primeros en describir en 1977 una glomerulopatía con depósitos de material fibrilar que se parecía al amiloide pero que no se teñía con el rojo Congo y que por inmunofluorescencia contenían IgG, IgM y C3. Duffy y col. (4) en su serie de ocho pacientes con esta patología glomerular utilizaron el término fibrilar para referirse a la ultraestructura de esos depósitos. Alpers y cols. (5) en 1987 adoptaron el nombre de «glomerulonefritis fibrilar» (GF) para esta nueva entidad; los depósitos eran extracelulares, estaban constituidos por fibrillas no ramificadas, orientadas al azar de aproximadamente 20 nm de diámetro y contenían inmunoglobulinas y complemento. Previamente, en 1980, Schwart y Lewis (6) habían descrito una glomerulopatía que tenía depósitos inmunes monoclonales IgG kappa con rasgos peculiares a nivel ultraestructural ya que presentaban morfología microtubular, eran de mayor tamaño, y se orientaban en bandas paralelas a la que denominaron «glomerulopatía inmunotactoide» (GIT). Casos adicionales con depósitos microtubulares mayores de 30 nm de diámetro han sido publicados posteriormente, siendo estas formas menos frecuentes que los casos con fibrillas menores.
La terminología usada en la literatura para esos patrones morfológicos ha sido confusa y existe un amplio debate sobre si la glomerulopatía fibrilar inmunotactoide representa una simple entidad (7-9) o son dos condiciones clínicopatológicas distintas (10). Korber y cols. han incluido bajo la designación de GIT a ambas variantes ultraestructurales de glomerulopatías idiopáticas argumentando que el término representa una contribución significativa al diagnóstico por incluir tanto morfopatología como composición de los depósitos sin que existan suficientes fundamentos clínicos o patogénicos para la subclasificación. Otros investigadores por el contrario mantienen la subdivisión de dichas glomerulopatías, GF y GIT, basadas en el tamaño de las fibrillas aduciendo importantes implicaciones clínicas (2,11,12). En opinión de Brady y cols. (13) sería más prudente adherirse a esa amplia terminología morfológica de «glomerulopatía fibrilar inmunotactoide» utilizada también por otros autores hasta que futuros estudios demuestren claras diferencias entre las variantes (14,15).
Formas Morfológicas
Microscopia óptica
Los hallazgos de microscopia óptica tanto en la GF como en la GIT son inespecíficos y variados. La expresión morfológica más frecuentemente observada es la de expansión mesangial y engrosamiento de la pared capilar que si no se acompañan de hipercelularidad pueden dar el aspecto de amiloidosis o de glomerulonefritis membranosa respectivamente. En otros casos cuando se asocian lesiones proliferativas con o sin semilunas la glomerulopatía fibrilar inmunotactoide puede tener apariencia de glomerulonefritis proliferativa focal o difusa, o de glomerulonefritis membranoproliferativa. Semilunas celulares han sido descritas en un 19-27% de los pacientes en diversas series (16,17), a veces afectando más del 50% de los glomérulos. En túbulos, intersticio o vasos no se evidencian lesiones específicas.
Los depósitos son PAS-positivos y negativos con plata metenamina, mostrando el mesangio en esta última técnica una típica apariencia moteada. Por definición los depósitos no se tiñen con rojo Congo o tioflavina-T para amiloide.
Inmunofluorescencia
El patrón de inmunofluorescencia corresponde a la distribución de los depósitos observados ultraestructuralmente. La apariencia más común es una fuerte tinción IgG difusa en paredes capilares y mesangio aunque también puede ser segmentaria. Los depósitos parietales pueden tener un aspecto granular tosco o acintado entrecortado relativamente típico que a veces puede imitar el patrón de la enfermedad por anticuerpos anti MBG y causar confusión diagnóstica especialmente si se acompaña de semilunas (18). En una minoría de las biopsias la tinción ha sido predominante o exclusivamente mesangial (16) y ocasionalmente negativa (19,20).
En los casos publicados de GF, más del 95% de las muestran eran positivas para IgG y menos frecuentes para IgM e IgA (60% y 30% respectivamente) que a su vez eran característicamente más débiles (1,5,6). Virtualmente todos los casos eran positivos para C3. Un estudio de Iskandar y cols. (16) analizando las subclases de IgG en pacientes con esa variante mostraba que la tinción estaba prácticamente restringida a la subclase IgG4. Ambas cadenas Kappa y Lambda eran detectadas en más del 75% de las GF. (16,17). La mayoría de los datos en la literatura indican que sólo entre el 10% y el 20% de los casos de GF tienen evidencia inmunohistológica para monoclonalidad, generalmente IgG kappa, en los depósitos fibrilares.
En la GIT también los depósitos más comunes han sido de IgG y C3, sin embargo la subclase IgG4 parece ser menos dominante, aunque son pocos los casos estudiados. Depósitos monoclonales, eran mucho más frecuentes en esta variante (2). Debe reseñarse que numerosos trabajos sobre glomerulopatía fibrilar inmunotactoide no contienen información sobre monoclonalidad de los depósitos.
Microscopia electrónica
El estudio de microscopia electrónica es esencial para el diagnóstico. Como ya se ha señalado pueden observarse dos patrones ultraestructurales basados en el tamaño, morfología y organización de las fibrillas en los depósitos glomerulares. La variante fibrilar está caracterizada por microfibrillas pequeñas que suelen tener un tamaño de 12 a 20 nm (<30 nm) y se disponen al azar; esas fibrillas son mayores que las del amiloide, que típicamente miden 6-10 nm de diámetro, y tienen aspecto sólido en los aumentos de rutina, sin embargo en grandes aumentos diversos autores han mostrado que las fibrillas de la GF al igual que las de amiloide pueden presentar luces (1,4,11). En la variante inmunotactoide las microfibrillas son más grandes (>30 nm), habitualmente tienen luces visibles (microtúbulos) y se organizan, al menos focalmente, en bandas paralelas (2). Los depósitos se localizan en el mesangio y en la mayoría de los casos también en las paredes capilares distribuidas en los espacios subendotelial y subepitelial así como a través de la membrana basal. Extenso borramiento de los pedicelos es un hallazgo frecuente (16,17). Aunque extremadamente raro han sido detectados depósitos fibrilares extraglomerulares focales en el intersticio rodeando capilares peritubulares y en membranas basales tubulares (7,21) e incluso depósitos extrarrenales en pulmón, hígado y médula ósea (22,23).
Debemos destacar que la subclasificación ultraestructural no está libre de solapamiento entre los dos patrones y en algunos pacientes han sido encontradas estructuras tubulares con diámetros inferiores a 30 nm (14,15). La relación entre monoclonalidad de los depósitos y tamaño y morfología fibrilar también es inconstante; aunque los depósitos monoclonales se asociaban más frecuentemente con estructuras microtubulares (>30 nm), también han sido descritos en casos de fibrillas de menor tamaño (14,15,24,25).
Se ilustran diversos aspectos morfológicos (microscopia óptica, inmunofluorescencia y microscopia electrónica) de dos casos con esta patología recogidos en nuestro servicio. El primero mostraba un típico patrón ultraestructural de GF (fig. 1). El segundo caso presentaba fibrillas que estaban densamente organizadas en bandas paralelas pero tenían un diámetro inferior a 30 nm y sólo en algunas de ellas se identificaban luces a grandes aumentos; para este caso, por compartir características de ambos patrones ultraestructurales, hemos preferido mantener el amplio término diagnóstico de «glomerulopatía fibrilar inmunotactoide» (fig. 2).

Fig. 1. Glomerulopatía
fibrilar. A) Glomérulo presentando expansión mesangial y engrosamiento de la
pared capilar. HE. B) Detalle del mesangio y de la pared capilar con material
PAS (+). C) Aspecto moteado del mesangio e irregularidades en la membrana basal.
Plata metenamina. D) Intensa positividad IgG parietal y mesangial difusa es
característica. Kappa y Lambda mostraban el mismo patrón. E) Microfotografía
electrónica. Membrana basal capilar infiltrada y reemplazada por fibrillas de
aproximadamente 15 nm, dispuestas al azar. POD, podocito: CAP. luz capilar.
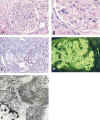
Fig. 2. Glomerulopatía
fibrilar inmunotactoide. A) Glomérulo con proliferación endocapilar
segmentaria y adherencias capsulares. HE. B) Detalle de la pared capilar
engrosada en otros segmentos. C) Semilunas celulares estaban presentes en el 50%
de los glomérulos. PAS D) Inmunofluorescencia. Intensa tinción de IgG parietal
y mesangial. E) Microfotografía electrónica. Depósitos subepiteliales,
subendoteliales y mesangiales, respetando la membrana basal capilar,
constituidos por fibrillas que aparecen densamente organizadas en bandas
paralelas; las fibrillas medían aproximadamente 15-20 nm, en las que se
evidenciaban ocasionales luces a grandes aumentos (recuadro). MBG, membrana
basal glomerular; POD, podocito; END, endotelio.
Patogénesis
La patogénesis de la glomerulopatía fibrilar inmunotactoide es desconocida. Los hallazgos inmunohistológicos indican que los depósitos patológicos fibrilares contienen inmunoglobulinas y complemento, hecho que confirmaron Yang y cols. (26) en microscopia inmunoelectrónica sugiriendo que las fibrillas representan componentes proteicos polimerizados.
Korbet y cols. (27) han propuesto que la uniformidad de la inmunoglobulina en los depósitos puede ser la base de las estructuras fibrilares. La homogeneidad de la inmunoglobulina monoclonal está clara en aquellos pacientes con GF que sólo tienen IgG kappa en los complejos inmunes; en aquellos pacientes con depósitos policlonales IgG sorprende que IgG 4 es la subclase exclusiva o predominante. En un estudio más reciente, Rostagno y cols. (28) sostienen la hipótesis de que un precursor sérico puede ser la fuente de los depósitos fibrilares y sugieren un papel de los complejos inmunoglobulin-fibronectina en la patogénesis de la GF. La recurrencia en los trasplantes es un argumento a favor de que los depósitos son derivados de proteínas circulantes.
Formas clínicas
La incidencia de glomerulopatía fibrilar inmunotactoide ha sido estimada en solo un 1% de las biopsias de riñón nativo (16,17). La variante inmunotactoide en su forma pura constituiría una minoría de los casos (0,2%) (14).
La presentación clínica es similar en las dos variantes con proteinuria elevada generalmente de rango nefrótico acompañada de hematuria y frecuentemente de hipertensión (13). Fallo renal rápidamente progresivo ha sido descrito en pacientes que presentaban semilunas o necrosis en más del 50% de los glomérulos. El 50% de los pacientes desarrollan enfermedad renal terminal a los 2-4 años del diagnóstico (5,7,9). Ha sido descrito un mejor pronóstico renal en la variante inmunotactoide (17). La enfermedad puede recurrir en el trasplante aunque no tiene porque llevar necesariamente a pérdida inmediata de la función renal (29).
La mayoría de los casos de glomerulopatía fibrilar inmunotactoide son idiopáticos (13,14), no obstante han sido descritas asociaciones con otras enfermedades como procesos linfoproliferativos (2,17,24), síndrome de Sjögren (17), vasculitis leucocitoclástica (30) o infección por hepatitis C (31). Ocasionalmente, en pacientes diagnosticados inicialmente de GIT ha sido detectada con posterioridad crioglobulinemia tipo II (2,31-32), esos hallazgos han sugerido que en algunos casos la glomerulopatía fibrilar inmunotactoide puede representar formas frustradas de crioglobulinemia (12,14).
Numerosos autores han señalado que pacientes con GIT presentan con una frecuencia extremadamente más alta procesos linfoproliferativos o disproteinemia/crioglobulinemia y que la distinción entre las dos variantes ayudaría potencialmente a identificar esos pacientes (10-12,17). Provonost y cols. (14) revisaron la incidencia de malignidad en 186 pacientes con glomerulopatía fibrilar inmunotactoide; cuando eran incluídos pacientes con una paraproteina circulante o en orina la incidencia de malignidad era significativamente más alta en los pacientes con GIT (33%) cuando se comparaba con la variante fibrilar (7%), en contraste cuando eran excluidos los enfermos con paraproteinemia la incidencia de malignidad era similar en ambas series (7%). Aunque se ha argumentado que el diagnóstico de glomerulopatía fibrilar inmunotactoide debe ser limitado a pacientes con enfermedad renal primaria excluyendo otras enfermedades sistémicas,1,9 muchos de los estudios publicados de GIT cumplen sólo parcialmente esos criterios e incluyen pacientes con gammapatías monoclonales y enfermedades linfoproliferativas (10,12,32).
Parece que puede haber razones morfológicas (ultraestructurales) para mantener la GF y la GIT como entidades clínicas distintas, presentando los pacientes con la variante fibrilar peor pronóstico renal y los pacientes con la variante inmunotactoide mayor probabilidad de tener o desarrollar enfermedades hematológicas. El problema es que cuantos más pacientes son identificados con esta patología, mayor es el solapamiento morfológico y algunos casos no encajan perfectamente en ninguna de las dos categorías (15,33). Serán necesarios estudios clinicopatológicos multicéntricos más amplios que demuestren diferencias clínicas específicas entre las variantes o que sean definidos diferentes mecanismos de fibrilogénesis para establecer conclusiones.
OTRAS ENFERMEDADES GLOMERULARES CON DEPÓSITOS FIBRILARES
Amiloidosis
La amiloidosis es la enfermedad con depósitos organizados mejor conocida. Ultraestructuralmente se caracteriza por presentar microfibrillas extracelulares de 8-10 nm de diámetro dispuestas al azar. Las principales proteínas de las fibrillas amiloides son AA (derivada de la proteína sérica amiloide A) y AL (derivada de inmunoglobulinas de cadenas ligeras). La reacción diagnóstica de las fibrillas amiloides con la tinción de rojo Congo, debida a una función de la estructura terciaria de la molécula de amiloide más que a la naturaleza de sus proteínas precursoras (9), es positiva en todas las formas de amiloidosis. La tinción rojo Congo separa la amiloidosis del resto de glomerulopatías con depósitos fibrilares que a continuación se comentan que son rojo Congo negativas.
Enfermedades con depósitos fibrilares no derivados de inmunoglobulínas
Diabetes Mellitus y enfermedades glomerulares esclerosantes
Ciertas enfermedades glomerulares esclerosantes, por ejemplo nefroangioesclerosis, glomeruloesclerosis segmentaria y focal o glomerulonefritis con esclerosis secundaria, pueden tener ocasionalmente fibrillas de 5 a 20 nm que suelen aparecer como pequeños acúmulos dispersos en el mesangio y generalmente no presentan problemas de diagnóstico diferencial (34).
En condiciones como diabetes mellitus, la expandida matriz mesangial puede tener en algunos casos una apariencia fibrilar, es la denominada «fibrilosis diabética» (fig. 3). Las fibrillas encontradas miden de 10 a 20 nm de diámetro, y no corresponden a depósitos inmunes a diferencia de la GF, sino a componentes normales o aberrantes de la matriz (11).

Fig. 3. Glomeruloesclerosis
diabética. La expandida matriz mesangial puede tener un aspecto fibrilar; es la
denominada "fibrilosis diabética" que no debe ser confundida con GF o
con amiloidosis.
Glomerulopatía colagenofibrótica
La glomerulopatía colagenofibrótica es una entidad rara recientemente definida caracterizada por depósito de fibras de colágeno en los glomérulos (35), puede ocurrir tanto en forma familiar como esporádica. La inmunotinción es negativa para inmunoglobulinas y complemento. A nivel ultraestructural las fibrillas de colágeno se localizan en mesangio y espacio subendotelial, miden hasta 100 nm (o más) de diámetro con típica periodicidad; no son difíciles de distinguir de las microfibrillas y microtúbulos de la GF y GIT debido a que aparecen curvadas y empaquetadas en bandas de variable grosor. El colágeno identificado ha sido de tipo I y III pero no tipo IV. Fibras de colágeno también han sido encontradas en la membrana basal engrosada del síndrome uña-rótula.
Glomerulopatía por fibronectina
La glomerulopatía por fibronectina es una enfermedad hereditaria descrita muy recientemente (36,37). Ultraestructuralmente presenta grandes depósitos electrodensos en el mesangio y espacio subendotelial que típicamente son finamente granulares aunque ocasionalmente pueden tener una subestructura fibrilar con un diámetro superponible al rango de la GF. La inmunotinción para fibronectina confirma el diagnóstico, presentando fuerte positividad en mesangio y paredes capilares con una relativa ausencia de depósito de inmunoglobulinas.
Enfermedades con depósitos fibrilares derivados de inmunoglobulinas
Crioglobulinemia
Han sido definidos tres tipos de crioglobulinas. El tipo I consiste en un simple componente de inmunoglobulina monoclonal, observada habitualmente en neoplasias hematolinfoides; el tipo II comprende dos componentes, una inmunoglobulina monoclonal, usualmente IgM kappa, con actividad antiglobulina (factor reumatoide) frente a una inmunoglobulina policlonal, detectada en infecciones, enfermedades autoinmunes, enfermedades crónicas hepáticas; y tipo III que es también una crioglobulina mixta en la que ambos componentes son policlonales. Los depósitos frecuentemente pero no siempre presentan configuración organizada. En el tipo I los depósitos pueden aparecer como bandas de fibrillas o túbulos; en la enfermedad crioglobulinémica tipo II tienen estructura tubular y en secciones transversas son anulares, generalmente menos definidas que en la GIT. Configuraciones tubulares también se han observado en el tipo III (38).
Lupus eritematoso sistémico
En la nefritis lúpica han sido descritos depósitos organizados tipo «fingerprint-like» (consistentes en líneas semicirculares oscuras y claras, cada una con un diámetro de 10-15 nm) en un 6-10 % de los pacientes. Recientemente Hvala y cols. (39) encontraban depósitos «fingerprint-like» en el 17% de su serie de 185 pacientes con LES, en 3 de los pacientes túbulos de 20-100 nm y en otros 2 pacientes fibrillas rojo congo negativas de 10 a 18 nm que simulaban túbulos y fibrillas de GIT y GF idiopáticas respectivamente, sin relación con crioglobulinas.
BIBLIOGRAFÍA
Korbet SM, Schwartz MM, Lewis EJ. The fibrillary glomerulopathies. Am J Kidney Dis 1994; 23: 751-65.
Alpers CE. Immunotactoid (microtubular) glomerulopathy: An entity distinct from fibrillary glomerulonephritis? Am J Kidney Dis 1992; 19: 185-91.
Rosenmann E, Eliakin M. Nephotic syndrome associated with amyloid-like glomerular deposits. Nephron 1977; 18: 301-8.
Duffy JL, Khurana E, Susin N, Gómez-León G, Churg J. Fibrillary renal deposits and nephritis. Am J Pathol 1983; 113: 279-90.
Alpers CE, Rennke HG, Hopper J, Biava CG. Fibrillary glomerulonephritis: an entity with unususal immunofluorescence features. Kidney Int 1987; 31: 781-9.
Schwartz MM, Lewis EJ: The quarterly case: nephrotic syndrome in a middle aged man. Ultrastruct Pathol 1980; 1: 575-82.
Korbet SM, Schwartz MM, Rosenberg BF, Sybley RK, Lewis EJ. Immunotactoide glomerulopathy. Medicine 1985; 64: 228.
Esparza A, Chazan J Ramakrishna N, NayaK N, Cavallo T. Fibrillary (immunotactoid) glomerulopathy- A posible role for kappa light chain in its etiology and/or pathogenesis. Am J Surg Pathol 1991; 15: 632-43.
Schwartz MM. Glomerular diseases with organized deposits. In Jennette JC, Olson JL, SchwartMM, Silva FG, eds. Heptinstall’s Pathology of the kidney, 5thed Philadelphia: Lippincott-Raven; 1998: 369-88.
Alpers CE. Fibrillary glomerulonephritis and immunotactoid glomerulopathy: two entities not one. Am J Kidney Dis 1993; 22: 488-451.
Jennette JC, Iskandar SWS, Falk RJ. Fibrillary glomerulonephritis, in Tisher CC, BrennerBM (eds): Renal pathology with clinical and functional correlation. 2end ed. Philadelphia, PA, Lippincott, 1994, pp. 553-63.
Ferrario F, Schiaffino E, Boeri R. Fibrillary and immunotactoid glomerulopathies. Renal Failure 1998; 20: 801-8.
Brady HR. Fibrillary glomerulopathy. Kidney Int 1998; 53: 1421-9.
Pronovost PH, Brady HR, Gunning ME, Espinoza O, Rennke HG. Clinical features predictors of disease progression and results of renal transplantation in fibrillary/immunotactoid glomerulopathy. Nephrol Dial Transplant 1996; 11: 837-42.
Ginneken E, Assmann K, Koolen M, Jansen J, Wetzels J. Fibrillary-immunotactoid glomerulopathy with renal deposits of IgA l; a rare cause of glomerulonephritis. Clin Nephrol 1999; 6: 383-9.
Iskandar S, Falk R, Jennette C. Clinical and pathology features of fibrillary glomerulonephritis. Kidney Int 1992; 42: 1401-7.
Fogo A, Nauman Q, Horn R. Morphologic and clinical features of fibrillary glomerulonephritis versus inmunotactoid glomerulopathy. Am J Kidney Dis 1993; 3: 367-77.
Sethi S, Adeyi O, Rennke HG. Un case of fibrillary glomerulonephritis with linear immunoglobulin G staining of the glomerular capillary walls. Arch Pathol Lab Med 2001; 125: 534-6.
Churg J, Venkataseshan S. Fibrillary Glomerulonephritis without immunoglobulin deposits in the kidney. Kidney Int 1993; 44: 837-42.
Vigil A, Oliet A, Gallar P, y cols. Rapidly progressive immunotactoid glomerulonephritis and multiple myeloma. Nephon 1998; 79: 238-40.
Adeyi A, Sethi S, Rennke H. Fibrillary glomerulonephritis: a report of 2 cases with extensive glomerular and tubular deposits. Human Pathol 2001; 32: 660-3.
Strom EH, Hurwitz N, Maryr A, y cols. Immunotactoid-like glomerulopathy with massive fibrillary deposits in liver and bone marrow in monoclonal gammopathy . Am J Nephrol 1996; 16: 523-8.
Da’as N, Kleinman Y, Polliack A, y cols. Glomerulopathy with massive bone marrow deposits in a patient with IgM Kappa monoclonal gammopathy D. of immunotactoid and hipocomplementemia. Am J Kidney Dis 2000; 2: 395-399. 17: 247-57.
Navarro-Antolin J, Quereda C, Mampaso F, y cols. Rapidly progressive fibrillary glomerulonephritis and cutaneus T-cell lymphoma. Nephron 1996; 73: 107-8.
Grove P, Neale P, Peck M, Schiller B, Haas M. Monoclonal immunoglobulin G1-Kappa fibrillary glomerulonephritis. Mod Pathol 1998; 11: 103-9.
Yang GCH, Nieto R, Stachura I, Gallo GR. Ultrastructural immunohistochemical localization of polyclonal IgG, C3 and amyloid P component on the Congo red-negative amyloid-like fibrils of fibrillary glomerulopathy. Am J Pathol 1992; 141: 409-19.
Korbet SM, Schwartz MM, Lewis EJ: Immunotactoid glomerulopathie. Am J Kidney Dis 1991; 17: 247-57.
Rostagno A, Vidal R, Kumar A, y cols. Fibrillary glomerulonephritis related to serum fibrillar immunoglobulin-fibronectin complexes. Am J Kidney Dis 1996; 28: 676-84.
Samaniego M, Nadasdy G, Laszik Z, Nadasdy T. Outcome of renal transplantation in fibrillary glomerulonephritis. Clin Nephrol 2001; 55: 159-66.
Orfila C, Meeus F, Bernadet P, Lepert JC, Suc JM. Immunotactoid glomerulopathy and cutaneous vasculitis. Am J Nephrol 1991; 11: 67-72.
Markowitz G, Cheng J, Colvin R, Trebbin W, D´Agati V. Hepatitis C viral infection is associated with fibrillary glomerulonephritis and immunotactoid glomerulopathy. J Am Soc Nephrol 1998; 9: 2244-52.
King J, Culppeper M, Corey R, Tucker A, Lajoie G, Howell D. Glomerulopathies with fibrillary deposits. Ultrastruct Pathol 2000; 24: 5-21.
Gibson IW, More IA. Glomerular pathology: recent advances. J Pathol 1998. 184: 123-9.
Kronz J, Neu A, Nadasdy T. When noncongophilic glomerular fibrils do not represent fibrillary glomerulonephritis: nonspecific mesangial fibrils in sclerosing glomeruli. Clin Nephrol 1998; 50: 218-23.
Ikeda K, Yokoyama H, Tomosugi N, Kida H, Ooshima A, Kobayashi K. Primary glomerular fibrosis: a new nephropathy caused by diffuse intraglomerular increase in atypical type III collagen fibres. Clin Nephrol 1990; 33: 155-9.
Mazzuco G, Maran E, Rollino c, Monga G. Glomerulonephritis with organized deposits: a mesangiopathic , not immune complex-mediated disease? A pathologic study of two cases in the same familly. Human Pathol 1992; 23: 63-8.
Strom EH, Banfi G, Krapf R, y cols. Glomerulopathy associated with predominant fibronectin deposits: a newly recognised hereditary disease. Kidney Int 1995; 48: 163-70.
Gallo G y Kumar A. Hematopoietic Disorders. In: Silva FG, Dágati VD, Nadasdy T eds. Renal biopsy interpretation. New YorK: Churchill Livingstone 1996: 259-82.
Hvala A, Kobenter T, Fergula D. Fingerprint and other organised deposits in lupus nephritis. Wien Klin Wochenschr 2000; 112: 711-5.
![]()